Linfohistiocitosis Hemofagocítica
en pediatría: Retos diagnósticos y terapéuticos en un hospital de tercer nivel
en Guayaquil, Ecuador.
Hemophagocytic lymphohistiocytosis in pediatrics:
Diagnostic and therapeutic challenges in a tertiary hospital in Guayaquil,
Ecuador.
González AR¹, Armijos
AN¹, Alvarado DG², Ramírez RR³, Campoverde LE³
1. Servicio de Pediatría, Hospital de Niños Dr.
Roberto Gilbert, Guayaquil, Ecuador.
2. Servicio de Pediatría, Instituto Oncológico
Nacional Dr. Juan Tanca Marengo (SOLCA), Guayaquil, Ecuador.
3. Servicio de Hematología Pediátrica, Hospital de
Niños Dr. Roberto Gilbert, Guayaquil, Ecuador.
González AR:
https://orcid.org/0000-0002-4289-3957
Armijos
AN: https://orcid.org/0000-0002-1768-0925
Alvarado DG: https://orcid.org/0000-0002-6354-2929
Ramírez RR: https://orcid.org/0000-0002-5125-9528
Campoverde LE: https://orcid.org/0009-0003-4825-8166
Palabras clave: Linfohistiocitosis hemofagocítica,
Ferritina, Dengue, COVID-19, Síndrome Inflamatorio Multisistémico Pediátrico.
Keywords: Hemophagocytic
Lymphohistiocytosis, Ferritin, Dengue, COVID-19, Pediatric Multisystem
Inflammatory Disease.
Resumen
Introducción: La
linfohistiocitosis hemofagocítica
(HLH) es un síndrome de hiperinflamación sistémica
grave, caracterizado por una activación incontrolada de linfocitos y macrófagos
que deriva en daño multiorgánico. En pediatría, la forma secundaria es la más
prevalente y suele ser desencadenada por infecciones, malignidades o
enfermedades autoinmunes. Debido a su rápida progresión y alta mortalidad, el
diagnóstico temprano es determinante para la supervivencia.
Objetivo:
Describir las características clínicas, hallazgos de laboratorio, etiología,
tratamiento y evolución de pacientes pediátricos con HLH en un hospital de
tercer nivel en Guayaquil, Ecuador.
Material y
Métodos: Estudio descriptivo y retrospectivo de una
serie de 22 casos (enero 2019 - septiembre 2025) diagnosticados bajo los
criterios HLH-2004. Se analizaron variables demográficas, paraclínicos,
estudios de médula ósea, desencadenantes infecciosos y protocolos terapéuticos.
Resultados: La
edad media fue de 4.26 años. La fiebre fue universal (100%) y la esplenomegalia
afectó al 86.36% de los pacientes. Se observó hiperferritinemia
universal (media: 8,804 ng/mL) e hipofibrinogenemia
en el 86.36% de los casos. Las infecciones virales fueron el principal
desencadenante (86.36%), destacando Dengue y SARS-CoV-2 (40.91% cada uno),
junto a una alta concurrencia de Síndrome Inflamatorio Multisistémico (PIMS) en
el 54.55%. El tratamiento incluyó dexametasona (90.91%), inmunoglobulina (54.55%)
y etopósido (18.18%). La mortalidad fue del 22.73%.
Discusión: La
HLH pediátrica presenta una alta morbilidad y se asocia frecuentemente a virus
endémicos y emergentes en la región. El diagnóstico basado en biomarcadores
como la ferritina es crucial, especialmente ante cuadros graves de Dengue o
PIMS, para iniciar un manejo inmunosupresor agresivo.
Abstract
Introduction: Hemophagocytic
lymphohistiocytosis (HLH) is a severe systemic hyperinflammatory syndrome
characterized by uncontrolled activation of lymphocytes and macrophages,
leading to multi-organ damage. In pediatrics, the secondary form is the most
prevalent and is usually triggered by infections, malignancies, or autoimmune
diseases. Due to its rapid progression and high mortality rate, early diagnosis
is crucial for survival.
Objective: To describe the clinical
characteristics, laboratory findings, etiology, treatment, and outcomes of
pediatric patients with HLH at a tertiary care hospital in Guayaquil, Ecuador.
Materials and
Methods: A descriptive and retrospective study of a series of 22 cases (January
2019–September 2025) diagnosed according to the HLH-2004 criteria. Demographic
variables, paraclinical findings, bone marrow studies, infectious triggers, and
treatment protocols were analyzed. Results: The mean age was 4.26 years. Fever
was universal (100%), and splenomegaly affected 86.36% of patients. Universal hyperferritinemia (mean: 8,804 ng/mL) and
hypofibrinogenemia were observed in 86.36% of cases. Viral infections were the
main trigger (86.36%), with dengue and SARS-CoV-2 being the most prevalent
(40.91% each), along with a high incidence of multisystem inflammatory syndrome
(MIS-C) in 54.55%. Treatment included dexamethasone (90.91%), immunoglobulin
(54.55%), and etoposide (18.18%). Mortality was 22.73%.
Discussion: Pediatric HLH presents
with high morbidity and is frequently associated with endemic and emerging
viruses in the region. Diagnosis based on biomarkers such as ferritin is
crucial, especially in severe cases of dengue or MIS-C, to initiate aggressive
immunosuppressive therapy.
Introducción
La linfohistiocitosis hemofagocítica
(HLH) es un síndrome de hiperinflamación sistémica
poco común, pero potencialmente mortal, caracterizado por una activación
incontrolada y proliferación de linfocitos y macrófagos, que resulta en una
respuesta inmunitaria desregulada y eventualmente daño multiorgánico (1).
Este síndrome representa un espectro de trastornos que, aunque raros, son
devastadores si no se diagnostican y tratan a tiempo. Se clasifica
principalmente en dos formas: la HLH primaria, de origen genético, y la HLH
secundaria, que es más común en la población pediátrica y puede ser
desencadenada por una variedad de condiciones subyacentes (2).
Entre los desencadenantes más frecuentes de la HLH secundaria se
encuentran las infecciones (especialmente virales), malignidades, enfermedades
autoinmunes (como la artritis idiopática juvenil sistémica) e
inmunodeficiencias (3). En la población pediátrica, la forma
secundaria es más común, a menudo desencadenada por virus como el Epstein-Barr
y Citomegalovirus (4). Dada su rápida progresión y la alta
mortalidad asociada si no se instaura un tratamiento oportuno, el
reconocimiento temprano de sus manifestaciones clínicas y de laboratorio es de
vital importancia para iniciar una terapia inmunosupresora y de soporte
agresiva (5). La naturaleza inespecífica y heterogenea
de su presentación pueden dificultar el diagnóstico, ya que a menudo es similar
a otras condiciones de alta prevalencia en pediatría, como sepsis, choque
séptico o enfermedades reumatológicas sistémicas (6).
La fisiopatología de la HLH implica una "tormenta de
citoquinas" debido a la activación y proliferación descontrolada de
linfocitos T y macrófagos, que no logran eliminar el estímulo desencadenante.
Esta desregulación inmunológica conduce a una producción excesiva de citoquinas
proinflamatorias, como la interleucina-6 (IL-6), el factor de necrosis tumoral
alfa (TNF-α) y el interferón gamma (IFN-γ), lo que resulta en daño tisular generalizado y eventualmente falla
multiorgánica (7). La HLH primaria se debe a mutaciones en genes que
afectan la función de las células T citotóxicas y las células Natural Killer
(NK) (8).
Los criterios diagnósticos establecidos por la Histiocyte
Society (HLH-2004) constituyen el estándar
internacional para la identificación de esta patología, representan una
revisión y actualización de los criterios HLH-1994, incorporando nuevos
biomarcadores y una comprensión más profunda de la fisiopatología de la enfermedad,
fueron desarrollados puesto que debido a la rareza y heterogeneidad clínica, a
menudo la misma era subdiagnosticada o diagnosticada tardíamente. Su implementación es crucial debido a la ausencia de
un signo patognomónico único; incluso la hemofagocitosis
en médula ósea puede no estar presente en estadios iniciales, y permiten un
diagnóstico temprano, lo cual es determinante para instaurar terapias de
rescate que mejoren la supervivencia de los pacientes (2,9). La hiperferritinemia, es en particular un biomarcador crucial,
con valores que superan lo que se observan en otras condiciones inflamatorias,
sirviendo como una señal de alerta temprana (10). Estudios
adicionales sobre las características clínicas y pronosticas de la HLH
secundaria en niños son importantes para optimizar el diagnóstico (11,12).
La elevación de Lactato Deshidrogenasa (LDH) y dímero D, junto con la hipofibrinogenemia, refuerzan el diagnóstico al reflejar la
citólisis y la coagulopatía de consumo tambien características
de este síndrome (13).
El tratamiento se basa en frenar la respuesta inmune y eliminar el
desencadenante; y se adaptan a la condición particular de cada paciente, los
protocolos actuales se basan predominantemente en el esquema HLH-94/2004, que
utiliza una combinación de corticoesteroides (como dexametasona),
quimioterápicos citotóxicos como el etopósido (para inducir la apoptosis de
linfocitos T activados) e inmunosupresores como la ciclosporina. En casos de
HLH secundaria, este manejo suele complementarse con inmunoglobulina intravenosa
(IVIG) y, más recientemente, con terapias biológicas dirigidas contra
citoquinas específicas (como el tocilizumab para la
IL-6), adaptando la intensidad del tratamiento a la gravedad y respuesta
clínica de cada paciente (14). La asociación con COVID-19 y el
Síndrome Inflamatorio Multisistémico Pediátrico (PIMS) es un hallazgo relevante
y emergente, que destaca la capacidad del SARS-CoV-2 para desencadenar
respuestas hiperinflamatorias graves en la población
pediátrica (15). Un análisis exhaustivo de la enfermedad y su manejo
es crucial para mejorar los resultados clínicos (16).
El presente estudio tiene como objetivo principal describir en
detalle las características clínicas, los hallazgos paraclínicos, el abordaje
terapéutico implementado y la evolución de una serie de casos de HLH en
pacientes pediátricos atendidos en el Hospital de Niños Dr. Roberto Gilbert de
Guayaquil, Ecuador, el cual funciona como un centro de referencia nacional de
tercer nivel de complejidad. A través de este análisis, buscamos contribuir al
conocimiento y optimización del manejo de esta compleja y desafiante patología,
proporcionando una visión más clara de su comportamiento en la población
pediátrica de nuestra región.
Materiales
y Métodos
Se realizó un análisis descriptivo y retrospectivo de 22 pacientes
pediátricos atendidos en el Hospital de Niños Dr. Roberto Gilbert de Guayaquil,
Ecuador. Se realizó una revisión de los registros electrónicos de pacientes que
presentaban diagnósticos relacionados con HLH (específicamente los códigos
D76.1 y D76.2 de la Clasificación Internacional de Enfermedades, 10ª Revisión -
CIE-10), durante el período comprendido entre enero de 2019 y septiembre de
2025.
Para cada paciente se
recolectaron datos demográficos, manifestaciones clínicas (fiebre,
hepatoesplenomegalia, exantema) y parámetros de laboratorio, incluyendo
hemograma, función hepática, renal y marcadores de hiperinflamación
(ferritina, LDH, dímero D y perfil de coagulación). La búsqueda etiológica se
realizó mediante cultivos convencionales, detección molecular por reacción en cadena
de la polimerasa y serología viral, mientras que el compromiso orgánico se
evaluó con radiografía de tórax, ecografía abdominal, tomografía y
ecocardiograma.
El diagnóstico se fundamentó en
los criterios de la Histiocyte Society
(HLH-2004), requiriendo la identificación de una mutación genética patogénica o
la presencia de al menos cinco de los ocho criterios estándar: 1) fiebre
persistente mayor o igual a 38.5 grados; 2) esplenomegalia mayor o igual a 3 cm
bajo el reborde costal o por imagen; 3) citopenias en al menos dos líneas
celulares (hemoglobina menor a 9 g/dL o menor a 12 g/dL en neonatos, plaquetas
menores a 100,000/uL y neutrófilos menores a 1,000/uL); 4) hipertrigliceridemia
mayor o igual a 265 mg/dL y/o hipofibrinogenemia
menor o igual a 150 mg/dL; 5) hemofagocitosis en
médula ósea, bazo o ganglios; 6) actividad de células NK reducida o ausente; 7)
ferritina mayor o igual a 500 ng/mL (valores
superiores a 3,000-10,000 ng/mL considerados más
específicos en pediatría) y 8) niveles de CD25 soluble (receptor de IL-2 alfa)
elevado. Cabe destacar que, debido a la falta de disponibilidad técnica en
nuestro medio local, no se realizaron estudios funcionales de degranulación celular (CD107a), expresión de perforinas, ni
estudios genéticos moleculares para mutaciones específicas (como STX11, UNC13D,
entre otros).
Se registraron los tratamientos
administrados (corticosteroides, inmunoglobulina IV, etopósido, ciclosporina, tocilizumab) y el desenlace clínico. El análisis
estadístico fue descriptivo, empleando medidas de tendencia central y
dispersión para variables cuantitativas, junto con frecuencias y porcentajes
para las cualitativas.
Resultados
Un total de 22 pacientes con
diagnóstico de HLH fueron incluidos en el presente estudio. La edad al momento
del diagnóstico mostró un rango amplio, desde los 2 meses hasta los 13 años y 9
meses, con una media de 4,26 +/- 2,98 años y una mediana de 2,62 años, lo que
sugiere una mayor prevalencia en las etapas pediátricas tempranas. En cuanto a
la distribución por sexo, se observó una predominancia masculina con 13 casos
(59,09%) frente a 9 casos femeninos (40.91%). Respecto a las comorbilidades, la
gran mayoría de los casos (90,91%) no presentaba patologías autoinmunes
previas; únicamente se documentó un caso de Artritis Idiopática Juvenil
Sistémica y uno de Púrpura Trombocitopénica Inmune.
En el perfil clínico, la fiebre fue
un hallazgo universal presente en el 100% de los pacientes, mientras que la
esplenomegalia se detectó en 19 casos (86,36%). Las alteraciones hematológicas
fueron consistentes con el síndrome de activación macrofágica:
la concentración media de hemoglobina fue de 7,98 +/- 2,16 g/dL, el recuento
promedio de plaquetas se situó en 84.955 +/- 61.727 por mm3 y la media de
neutrófilos fue de 1.970 ± 2.460 por mm3, evidenciando citopenias graves en
múltiples líneas. Los marcadores de hiperinflamación
mostraron elevaciones extremas; la ferritina sérica alcanzó una media de 8.804
+/- 12.710 ng/mL, destacando que un 68.18% de los pacientes
superó el umbral de 3.000 ng/mL y un 27,27% sobrepasó
los 10.000 ng/mL, con un valor máximo de 50,283 ng/mL (Tabla 1).
Tabla 1:
Características Demográficas, Clínicas, Hallazgos de Laboratorio y de Aspirado
de Médula Ósea (PAMO) al Diagnóstico.
|
Variable |
n (%) o Valor |
Media ± DE |
Mediana (Rango) |
|
Características Demográficas |
|||
|
Edad
(años) |
22 (100%) |
4,26±2,98 |
2,62 (0,17 - 13,75) |
|
Sexo (Masculino) |
13 (59,09%) |
- |
- |
|
Manifestaciones Clínicas |
|||
|
Fiebre |
22 (100%) |
- |
- |
|
Esplenomegalia |
19 (86,36%) |
- |
- |
|
Parámetros Hematológicos |
|||
|
Hemoglobina
(g/dL) |
22 (100%) |
7,98±2,16 |
8,50 (3,60 - 13,20) |
|
Plaquetas (×103/mm3) |
22 (100%) |
84,96±61,73 |
76,00 (6,00 - 220,00) |
|
Neutrófilos
(×103/mm3) |
22 (100%) |
1,97±2,46 |
1,09 (0,03 - 7,96) |
|
Biomarcadores de Inflamación |
|||
|
Ferritina
(ng/mL) |
22 (100%) |
8.804,8±12.710 |
4.842 (849 - 50.283) |
|
Ferritina ≥3.000 ng/mL |
15 (68,18%) |
- |
- |
|
Ferritina
≥10.000 ng/mL |
6 (27,27%) |
- |
- |
|
Triglicéridos (mg/dL) |
22 (100%) |
290,0±164,7 |
271 (72 - 698) |
|
Fibrinógeno
(mg/dL)* |
20 (90,91%) |
145,4±109,1 |
117 (50 - 506) |
|
Dímero D (mcg/mL) |
21 (95,45%) |
5,79±3,74 |
5,70 (0,77 - 12,50) |
|
Disfunción de Órgano /
Citólisis |
|||
|
LDH (U/L) |
22 (100%) |
1.570,6±1.164 |
1.365 (344 - 4.522) |
|
AST
(U/L) |
22 (100%) |
1.100,8±2.629 |
390,5 (21 - 11.902) |
|
ALT (U/L) |
22 (100%) |
495,7±808,7 |
228 (28 - 3.155) |
|
Bilirrubina
Total (mg/dL) |
20 (90,91%) |
1,77±2,16 |
0,80 (0,27 - 8,45) |
|
Albúmina (g/dL) |
22 (100%) |
2,86±0,57 |
2,70 (2,15 - 4,40) |
|
Estudios de Médula Ósea |
|||
|
PAMO realizada |
16 (72,73%) |
- |
- |
|
Hemofagocitosis confirmada** |
13 (81,25%) |
- |
- |
Nota: DE:
Desviación estándar; PAMO: Punción aspiración de médula ósea. *En 2 casos
(9,09%) no se obtuvo valor numérico de fibrinógeno por ausencia de coagulación
de la muestra (consumo severo). **Porcentaje calculado sobre el total de PAMO
realizadas (n=16).
La coagulopatía y el compromiso
metabólico fueron significativos. El fibrinógeno promedio fue de 145,45 +/- 109,11
mg/dL, presentándose hipofibrinogenemia en el 86.36%
de los casos; es notable que en dos pacientes la coagulopatía fue tan severa
que la muestra no fue coagulable. La hipertrigliceridemia
afectó al 50% de la cohorte con una media de 290,05 mg/dL. Los indicadores de
daño orgánico revelaron una alta carga de enfermedad, con una LDH promedio de
1.570,68 U/L y una elevación marcada de transaminasas con una Aspartato
Aminotransferasa (AST) media de 1,100.82 U/L y una y Alanina Aminotransferasa (ALT)
media de 495,73 U/L). La hipoalbuminemia fue casi constante (media 2,86 g/dL) y
se observó una tendencia a la hiponatremia. En el estudio de extensión, la hemofagocitosis fue confirmada mediante punción aspiración
de médula ósea (PAMO) en el 81,25% de los 16 pacientes en quienes se realizó el
procedimiento (Imagen 1).
Cabe señalar que la determinación de
la actividad de las células Natural Killer (NK) y los niveles de CD25 soluble
(receptor de IL-2 alfa) no se realizaron, debido a la falta de disponibilidad
técnica de estas pruebas en nuestro centro durante el periodo de estudio (Tabla 2).
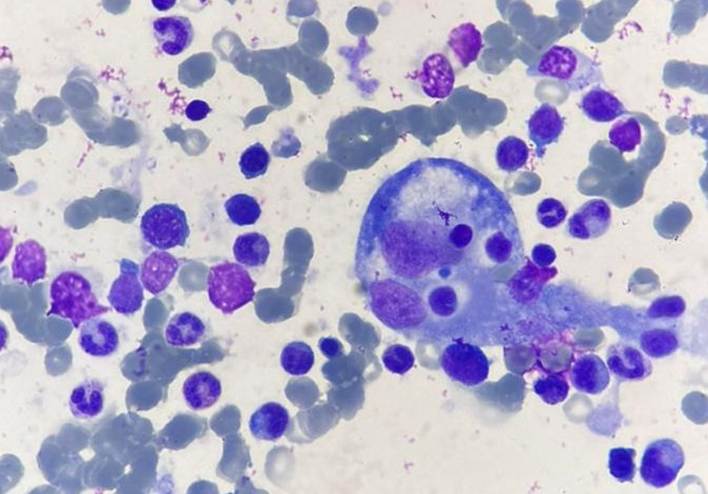
Imagen 1: PAMO con hemofagocitosis, se observa un Macrófago que ha fagocitado
varios eritrocitos y plaquetas. Tinción de Wright-Giemsa, Micrografía tomada a
1000x. Imagen cortesía del Dr. Robinson Ramírez Ruiz.
Tabla 2:
Cumplimiento de los criterios diagnósticos HLH-2004
|
Criterio Diagnóstico
(HLH-2004) |
Definición |
Pacientes (n) |
Porcentaje (%) |
|
Fiebre |
≥38,5∘C |
22 |
100% |
|
Esplenomegalia |
Hallazgo clínico o por imagen |
19 |
86,36% |
|
Citopenias (≥2 líneas) |
Hb <9 g/dL, Plt
<100.000/mm3, Neutrófilos <1.000/mm3 |
13 |
59,09% |
|
Hipertrigliceridemia y/o Hipofibrinogenemia |
Trig. ≥265 mg/dL y/o Fibrinógeno
≤150 mg/dL |
20 |
90,91% |
|
Hemofagocitosis |
Evidencia en médula ósea, bazo o ganglio |
13 |
59,09%* |
|
Hiperferritinemia |
≥500 ng/mL |
22 |
100% |
|
Actividad NK baja/ausente |
Mediante ensayo de lisis celular |
N/A |
0% |
|
CD25 soluble elevado |
≥ 2.400 U/mL |
N/A |
0% |
Nota: Hb:
hemoglobina; Plt:
plaquetas; Neut:
neutrófilos; Trig: triglicéridos;
NK: células natural killer; CD25:
receptor de interleucina-2 alfa soluble. N/A: No disponible en el centro. *Porcentaje
calculado sobre el total de la cohorte (n=22).
Desde el punto de vista etiológico,
se identificó un desencadenante viral en el 86.36% de los casos, destacando el
Dengue y el SARS-CoV-2 (40,91% cada uno) y el virus de Epstein-Barr (31,82%),
Las infecciones bacterianas también fueron significativas, identificándose al
menos una etiología bacteriana en 13 de los 22 pacientes (59,09%). Los
patógenos bacterianos más frecuentes fueron Salmonella enterica
y Escherichia coli Betalactamasas
de Espectro Extendido (BLEE) con un 13,64% cada uno, seguidos por Staphylococcus aureus en un
9,09% (Tabla 3).
Tabla 3:
Etiologías Infecciosas Identificadas
|
Paciente |
Etiología viral principal |
Otras etiologías virales |
Etiología bacteriana principal |
Otras etiologías bacterianas |
|
1 |
Epstein Barr |
No |
No |
No |
|
2 |
No |
No |
Stenotrophomona Maltophila |
No |
|
3 |
Dengue |
SARS-CoV-2, Hepatitis A |
No |
No |
|
4 |
Parvovirus B19 |
CMV, VSR |
Klebsiella ozaenae
BLEE |
No |
|
5 |
Epstein Barr |
SARS-CoV-2, Dengue |
Salmonella enterica |
No |
|
6 |
Dengue |
No |
No |
No |
|
7 |
Epstein Barr |
Parvovirus B19 |
Escherichia coli BLEE |
No |
|
8 |
SARS-CoV-2 |
No |
No |
No |
|
9 |
SARS-CoV-2 |
Epstein Barr, CMV |
Escherichia coli BLEE |
Streptococcus viridans |
|
10 |
SARS-CoV-2 |
No |
Salmonella
Spp. |
Escherichia coli |
|
11 |
No |
No |
Salmonella Spp. |
Serratia marcescens, Leptospira |
|
12 |
CMV |
SARS-CoV-2 |
Staphylococcus aureus |
No |
|
13 |
CMV |
SARS-CoV-2 |
No |
No |
|
14 |
Dengue |
SARS-CoV-2 |
No |
No |
|
15 |
Dengue |
CMV |
No |
No |
|
16 |
Epstein Barr |
No |
Pseudomonas
aeruginosa |
No |
|
17 |
Dengue |
SARS-CoV-2 |
Escherichia coli BLEE |
No |
|
18 |
Dengue |
Epstein Barr, Rotavirus |
Salmonella
enterica |
No |
|
19 |
Dengue |
Epstein Barr, VSR |
Staphylococcus aureus |
No |
|
20 |
No |
No |
Salmonella
enterica |
No |
|
21 |
Dengue |
No |
No |
No |
|
22 |
Dengue |
No |
No |
No |
Nota: VSR: Virus
Sincitial Respiratorio. CMV: Citomegalovirus
Las complicaciones sistémicas fueron
frecuentes y graves: neumonía (63,64%), fallo hepático (54,55%), hemorragias
(63,64%) falla multiorgánica (36,36%), y Síndrome de Distrés Respiratorio Agudo
(27,27%). También se registraron derrames en cavidades serosas, con derrame
pleural bilateral (54,55%), pericárdico (40,91%) y ascitis (36,36%). Es de
notar que el dengue grave fue identificado como una condición asociada en 5
pacientes (22,73%), sugiriendo un posible factor desencadenante o una
superposición de síndromes en algunos casos. Se resalta que el 54,55% de los
pacientes presentó criterios concurrentes para Síndrome Inflamatorio
Multisistémico Pediátrico (PIMS).
El manejo terapéutico incluyó
dexametasona en el 90.91% de los pacientes, inmunoglobulina intravenosa en el
54,55% y ciclosporina en el 27,27%. El etopósido fue administrado en el 18,18%
de los casos y solo un paciente recibió tocilizumab.
La estancia hospitalaria promedio fue de 22 días (rango 6 a 61 días). La
mortalidad global fue del 22,73% (5 pacientes); de estos fallecimientos, el 60%
se atribuyó directamente a complicaciones de la HLH y el 40% restante a sepsis
secundaria grave por gérmenes como Staphylococcus aureus (Tabla 4).
Tabla 4: Complicaciones,
Co-morbilidades y Mortalidad
|
Variable |
n (%) o Valor |
Media
± DE / Mediana (Rango) |
|
Complicaciones Clínicas |
||
|
Neumonía |
14 (63,64%) |
- |
|
Manifestaciones de sangrado |
14 (63,64%) |
- |
|
Fallo hepático |
12 (54,55%) |
- |
|
Derrame pleural bilateral |
12 (54,55%) |
- |
|
Choque séptico |
9 (40,91%) |
- |
|
Derrame pericárdico |
9 (40,91%) |
- |
|
Falla multiorgánica |
8 (36,36%) |
- |
|
Ascitis |
8 (36,36%) |
- |
|
Sepsis |
7 (31,82%) |
- |
|
CID |
6 (27,27%) |
- |
|
SDRA |
6 (27,27%) |
- |
|
Factores Asociados / Co-morbilidades |
||
|
PIMS concurrente |
12 (54,55%) |
- |
|
Dengue grave |
5 (22,73%) |
- |
|
Evolución y Desenlace |
||
|
Estancia hospitalaria (días), media ±
DE |
- |
22,09±11,75 |
|
Mediana (rango) de estancia |
- |
18,5 (6 - 61) |
|
Mortalidad Global |
5 (22,73%) |
- |
|
Causas de Fallecimiento
(n=5) |
||
|
Complicaciones directas de HLH |
3 (60,00%) |
- |
|
Septicemia* |
2 (40,00%) |
- |
Nota: CID:
Coagulación intravascular diseminada; SDRA: Síndrome de distrés respiratorio
agudo; PIMS: Síndrome inflamatorio multisistémico pediátrico. *Incluye un caso
de septicemia no especificada y un caso por Staphylococcus aureus.
Discusión
La HLH en la población pediátrica representa un síndrome de hiperinflamación severa con una presentación clínica
heterogénea y un pronóstico sombrío si no se diagnostica y trata apropiadamente.
Nuestra serie de 22 casos de HLH pediátrica en un centro de atención terciaria
refleja esta complejidad, destacando hallazgos consistentes con la literatura
internacional, pero también particularidades relevantes para nuestra población.
La distribución de edad en nuestra cohorte, con una edad media de 4,26
años, sugiere que la HLH se presenta con mayor frecuencia en la primera
infancia, un patrón observado en otros estudios pediátricos (1,6).
La predominancia masculina, con un 59,09% de los casos, también está en línea
con las tendencias reportadas para esta enfermedad. La presentación clínica
inicial fue clásica, con fiebre persistente como un hallazgo universal y esplenomegalia
en la gran mayoría (86,36%) de los pacientes, signos que deben levantar una
alta sospecha clínica (2, 6).
Los parámetros de laboratorio de nuestra cohorte reflejaron el
estado hiperinflamatorio y el síndrome de consumo
característicos de la enfermedad. La hiperferritinemia,
con una media elevada, se destacó como un marcador clave y un signo de alerta
crítico, superando ampliamente lo reportado en otras condiciones inflamatorias
(10). Las citopenias, incluida la anemia y la trombocitopenia, fueron
hallazgos constantes. Además, los marcadores de disfunción hepática (elevación
de AST/ALT, hiperbilirrubinemia), citólisis (elevación de LDH) y coagulopatía (hipofibrinogenemia, elevación de Dímero D) fueron
notablemente prevalentes y severos, en concordancia con la fisiopatología de la
HLH (13).
La hemofagocitosis en la médula ósea se
identificó en un alto porcentajes de pacientes a quienes se les realizó una
PAMO (81,25%), sin
embargo, no fue un hallazgo universal. En un
27,3% de los pacientes (n=6), la inestabilidad clínica impidió realizar la PAMO, priorizándose la estabilización ante
niveles críticos de ferritina e infecciones confirmadas. Aunque el inicio de esteroides sin descartar procesos linfoproliferativos
es una debilidad diagnóstica por el riesgo de enmascararlos, la evolución favorable y la ausencia de estos en el
seguimiento validan retrospectivamente este abordaje clínico. Este hallazgo reitera que su ausencia no excluye el diagnóstico
de HLH, como ha sido ampliamente reconocido (2).Una limitante
importante fue no contar con otros métodos diagnósticos o criterios como medición
de activadad NK o de CD 25, una realidad compartida
con muchos otros centros de la región.
Un hallazgo particularmente relevante de nuestra serie es la alta
frecuencia de agentes infecciosos como desencadenantes. Se identificó una
etiología viral en el 86.36% de los pacientes, con el Dengue y el SARS-CoV-2
siendo los virus más prevalentes (13). La coexistencia de HLH y
Dengue representa un desafío diagnóstico debido al mimetismo clínico y
analítico entre ambas entidades (fiebre, citopenias, falla hepática). Es
importante mantener un alto índice de sospecha en áreas endémicas para evitar
el subdiagnóstico de HLH en pacientes con dengue
grave que presenta una evolución tórpida. La
significativa presencia de SARS-CoV-2 y la concurrencia de Síndrome
Inflamatorio Multisistémico (PIMS) en más de la mitad de los pacientes (54,55%),
resaltan la capacidad del virus para provocar una respuesta hiperinflamatoria
extrema, un área de investigación emergente, luego de la pandemia (6,15).
Las infecciones bacterianas también jugaron un rol importante, identificándose ampliamente
y subrayando la necesidad de un enfoque diagnóstico microbiológico exhaustivo. Si bien la mayoría de los casos se presentaron en un contexto
epidemiológico de arbovirosis y pandemia por
SARS-CoV-2, la imposibilidad de realizar un tamizaje genético exhaustivo impide
descartar de forma absoluta la presencia de formas primarias desencadenadas por
estos virus. Por tanto, el carácter secundario de la cohorte se asume basándose
en la asociación temporal y clínica, reconociendo esta limitación en el estudio
de los pacientes.
La variabilidad en el manejo terapéutico de nuestra cohorte refleja
la necesidad de adaptar el tratamiento, si bien la Dexametasona fue el pilar
del tratamiento inicial, el uso de etopósido se limitó solo a un grupo de los pacientes.
Esta subutilización, refleja la tendencia a reservar agentes quimioterapéuticos
para casos refractarios o de mayor gravedad en contextos de HLH secundaria a
virus (2,14). El uso excepcional de tocilizumab
nos señala que su administración se reserva para escenarios refractarios, o
bien como una estrategia de rescate terapéutico. A pesar de estas variaciones
terapéuticas y las limitaciones diagnósticas, la mortalidad del 22,73% en
nuestra serie es comparable a lo reportado en la literatura internacional,
subrayando la eficacia del manejo inmunosupresor instaurado basado en la
sospecha clínica temprana (9,11).
Conclusiones
La linfohistiocitosis hemofagocítica
en la población pediátrica ecuatoriana está íntimamente ligada a etiologías
virales endémicas y emergentes. Ante la ausencia de pruebas diagnósticas
avanzadas como la actividad NK, el uso riguroso de criterios clínicos y
biomarcadores accesibles, particularmente la ferritina superior a 3,000 ng/mL, es determinante para la supervivencia. Es imperativo
mantener un alto índice de sospecha en pacientes con dengue grave o PIMS que
presenten una evolución tórpida, para garantizar un enfoque terapéutico
agresivo y multidisciplinario.
Agradecimientos
Los autores desean expresar su más sincero agradecimiento al
personal médico, de enfermería y de apoyo del Hospital de Niños Dr. Roberto
Gilbert.
Declaración de conflictos de interés: los
autores declaran no poseer conflictos.
Referencias
1.
Jordan MB, Allen CE, Weitzman S, Filipovich AH,
McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011; 118(15):4041-52.
2.
Henter
JI, Horne A, Aricó M y col. HLH-2004:
Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood
Cancer. 2007; 48(2):124-31.
3.
Toumeh N, Abu-Zeinah K, Godby R. Hemophagocytic
lymphohistiocytosis (HLH): a narrative review of the pathogenesis, clinical
presentation, diagnosis, treatment, and prognosis. Ann
Blood. 2025; 10.
4.
Filipovich AH. Hemophagocytic
lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008; 28(2):293-viii.
5.
Wang Y, Wang Z. Treatment of hemophagocytic
lymphohistiocytosis. Curr Opin Hematol. 2017;
24(1):54-8.
6.
Canna SW, Marsh RA. Pediatric hemophagocytic
lymphohistiocytosis. Blood. 2020; 135(16):1332-43.
7.
Schulert GS, Grom AA.
Pathogenesis of macrophage activation syndrome and potential for
cytokine-directed therapies. Annu Rev Med. 2015;
66:145-59.
8.
Zhao C, Zhang Q, Zhang R y col. Genetic and
clinical characteristics of primary hemophagocytic lymphohistiocytosis in
children. Ann Hematol. 2024; 103(1):17-28.
9.
Terán-Ibarra
FS y col. Características clínicas de la linfohistiocitosis
hemofagocítica en un hospital universitario de tercer
nivel en Bogotá, Colombi. Infectio. 2024;
28(3):139-44.
10.
Al-Samkari H,
Berliner N. Hemophagocytic lymphohistiocytosis. Annu
Rev Pathol. 2018; 13:27-49.
11.
Abbasi AM, Shaikh MU, Shariq M y col. Outcome
of patients with primary and secondary hemophagocytic lymphohistiocytosis: A
retrospective analysis from a tertiary care center. Medicine
(Baltimore). 2023; 102(43):e34898.
12.
Bergsten
E, Horne A, Aricó M y col. Confirmed
efficacy of etoposide and dexamethasone in HLH treatment: long-term results of
the cooperative HLH-2004 study. Blood. 2017;
130(25):2728-38.
13.
Rosado FG, Kim AS. Hemophagocytic
lymphohistiocytosis: an update on diagnosis and pathogenesis. Am J Clin Pathol. 2013; 139(6):713-27.
14.
Ehl S, Astigarraga I,
von Bahr Greenwood T y col. Recommendations for the use of etoposide-based
therapy and bone marrow transplantation for the treatment of HLH: consensus
statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Immunol Pract. 2018; 6(5):1508-17.
15.
Ombrello MJ, Schulert GS. COVID-19 and cytokine storm syndrome: are
there lessons from macrophage activation syndrome? Transl Res. 2021; 232:1-12.
16. George MR.
Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014; 5:69-86.