Síndrome asociado a MECOM: insuficiencia medular grave sin anomalías esqueléticas, como variante clínica distinta. Reporte de caso.
MECOM-associated syndrome presenting as severe bone marrow failure without skeletal abnormalities: a distinct clinical variant. Case report.
Arenas Contreras EJ¹, Bárcenas Narváez WA²
1. Residente de Pediatría, Universidad del Norte, Barranquilla, Colombia. ORCID: https://orcid.org/0009-0006-6389-1121
2. Especialista en Hemato Oncología pediátrica. Universidad del Norte. Barranquilla, Colombia. ORCID: https://orcid.org/0000-0003-3001-0253
Palabras clave: MECOM,
insuficiencia medular,
pancitopenia,
fallo medular.
Keywords: MECOM,
bone marrow failure,
pancytopenia.
Resumen
El síndrome asociado a MECOM abarca un espectro de alteraciones hematológicas y esqueléticas, siendo la insuficiencia medular grave una de sus principales manifestaciones. Se presenta el caso de un lactante masculino de siete meses que debutó a los dos meses de vida con pancitopenia severa hasta insuficiencia medular. Los estudios de médula ósea evidenciaron hipocelularidad con escasa hematopoyesis y megacariocitos hipolobulados, con cariotipo normal (46, XY). El exoma clínico identificó una nueva variante heterocigótica en el gen MECOM, clasificada como probablemente patogénica. Las radiografías descartaron sinostosis radiocubital u otras alteraciones óseas, ampliando el espectro geno-fenotípico de las variantes del MECOM.
Abstract
The MECOM-associated syndrome encompasses a spectrum of hematologic and skeletal abnormalities, with severe bone marrow failure being one of its main manifestations. The case of a seven-month-old male infant, presenting at two months of age with severe pancytopenia, is described. Bone marrow studies revealed hypocellularity with poor hematopoietic representation and hypolobulated megakaryocytes, with a normal karyotype (46, XY). Clinical exome sequencing identified a novel heterozygous variant in the MECOM gene, classified as likely pathogenic. Radiographs ruled out radioulnar synostosis or other skeletal abnormalities, thereby expanding the geno-phenotypic spectrum of MECOM variants.
Introducción
El síndrome asociado a MECOM hace parte de los diagnósticos diferenciales de fallos medulares congénitos. Se caracteriza por aparición en edades tempranas, como neonatos y lactantes, con compromiso principalmente hematológico y óseo. No obstante, se han reportado compromisos extramedulares, entre los que se encuentran: malformaciones renales, cardíacas, craneofaciales, hasta compromiso auditivo y retraso en el desarrollo(1).
El gen MECOM (MDS1 and EVI1 Complex Locus) se ubica en el cromosoma 3q26.2, codificando factores de transcripción MDS1, MDS1-EVI1 y EVI1 (ecotropic viral integration site 1), que regulan el desarrollo embrionario y la renovación de células madre hematopoyéticas, así como la diferenciación megacariocítica y el desarrollo de hueso y cartílago(2). Estas proteínas contienen dedos de zinc, que median la unión a ADN y reclutamiento de cofactores críticos del ciclo celular, proliferación y mantenimiento, necesarios para la autorenovación de células madre hematopoyéticas y su diferenciación(3,4). La pérdida de función conduce a la represión de estos genes, generando un espectro de compromiso hematológico que varía desde pancitopenia severa de inicio temprano, hasta formas asociadas a haploinsuficiencia, caracterizadas por trombocitopenia inicial que puede progresar en semanas o meses a pancitopenia, en el contexto de disfunción celular subyacente(5). De esta manera, en la actualización de 2022 del Comité de Expertos de la Unión Internacional de Sociedades Inmunológicas, la deficiencia de MECOM se incluyó como un error innato de la inmunidad en la categoría de falla de médula ósea(5). Por otro lado, en mutaciones con ganancia de función como inv(3) (q21q26) o t(3;3) (q21;126) puede actuar como oncogén, con proliferación aberrante de los factores, progresando a leucemia mieloide aguda (LMA) de mal pronóstico(4,5).
Como se mencionó anteriormente, el espectro clínico es amplio, dado por las múltiples mutaciones heterocigóticas de MECOM, generando una enfermedad sindrómica con un patrón fenotípico variable. De esta manera, aunque el compromiso hematológico y óseo son las manifestaciones más comunes, ninguna de estas manifestaciones clínicas son obligatorias para el diagnóstico(1).
En este informe se presenta un caso de síndrome asociado a MECOM con insuficiencia medular grave sin compromiso óseo, con mutación única, no descrita previamente en la literatura.
Caso clínico
Lactante masculino de 7 meses de edad, natural del corregimiento de Caracolí (Atlántico, Colombia), nacido de término con peso y talla adecuados para la edad gestacional, sin antecedentes perinatales. Entre los antecedentes familiares se destaca que la madre y la abuela materna son portadoras del rasgo de anemia de células falciformes. No hay antecedentes de consanguinidad ni de enfermedades hematológicas malignas conocidas.
A los dos meses de vida, el paciente inicia cuadro de hematoquecia, motivo por el cual se realizan estudios de laboratorio que evidencian pancitopenia severa (hemoglobina 5 g/dL, hematocrito 16.2%, glóbulos rojos 1.67*106 µL, volumen corpuscular medio 97 fL, hematocrito corpuscular medio 34 pg, amplitud de distribución eritrocitaria 15.8%, leucocitos 4.870/µL, neutrófilos 13%, linfocitos 80.1%, monocitos 6.9% y plaquetas 17.000/µL), con indicación de transfusión de glóbulos rojos. Por la gravedad del compromiso hematológico, es remitido a un centro de mayor complejidad para valoración y manejo integral, siendo hospitalizado en Unidad de Cuidados Intensivos pediátricos.
Durante su estancia hospitalaria, se plantearon impresiones diagnósticas entre las que se encontraban: trombocitopenia inmune, síndrome de Evans, anemia de Fanconi y síndrome mielodisplásico. Se descartaron infecciones congénitas del grupo TORCH, y tanto la ecografía abdominal como el ecocardiograma Doppler color fueron reportados dentro de límites normales.
A los tres meses de edad, se realizó aspirado y biopsia de médula ósea con citometría de flujo hipocelular, que mostró una población de linfocitos B inmaduros aberrantes (0,08%). No obstante, la biopsia inicial no evidenció hallazgos patológicos significativos, no se identificaron poblaciones celulares que expresaran inmunofenotipo sospechoso de neoplasia, motivo por el cual se repitió el estudio al mes, encontrando tanto en citometría de flujo como biopsia, reporte de médula ósea hipocelular con celularidad del 10%, escasa representación de las líneas hematopoyéticas y megacariocitos hipolobulados. El cariotipo en médula ósea (46, XY [20]), fue normal, sin alteraciones numéricas ni estructurales.
Durante su evolución presentó infecciones recurrentes, incluyendo infecciones urinarias (Pseudomonas aeruginosa, Proteus mirabilis, Enterococcus faecium), bacteriemia por Pseudomonas aeruginosa, neumonías e infecciones de piel y tejidos blandos, que requirieron manejo antimicrobiano prolongado.
Debido a la refractariedad del compromiso hematológico (Figura 1), pese al tratamiento con corticoides, inmunoglobulina intravenosa, ácido fólico, vitamina B12, ácido ascórbico y eltrombopag, se amplió el estudio etiológico para descartar fallo medular, por lo que se solicitó exoma clínico, que identificó una nueva mutación heterocigótica en el gen MECOM (NM_004991.4: c.2242 C>T; p. Arg 748** ubicada en el exón 8/17, generando un codón de parada prematuro. Esta variante no estaba reportada en bases de datos internacionales (dbSNP, ClinVar) ni en la literatura médica, y fue clasificada como probablemente patogénica según los criterios del Colegio Americano de Genética Médica y Genómica (ACMG) (Tabla 1).
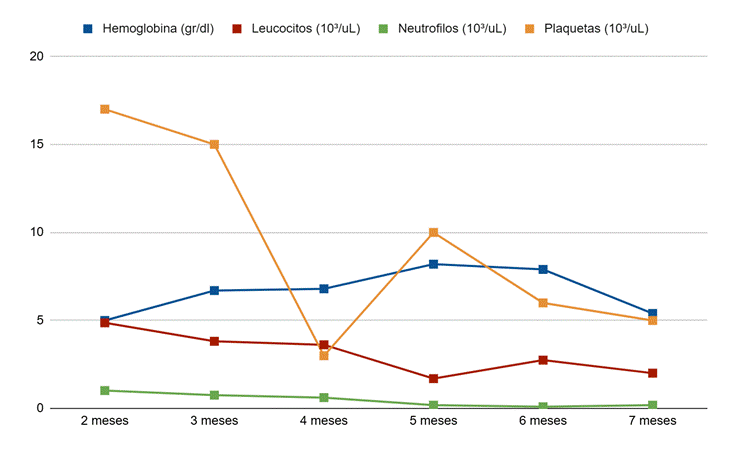
Figura 1. Evolución seriada de parámetros hematológicos del paciente.
|
Gen |
Variante |
Cigosidad |
Clasificación |
Fenotipo |
|
MECOM NM_004991.4 |
c.2242C>T p.Arg748* |
Heterocigoto |
Probablemente Patogénica |
Sinostosis radiocubital con trombocitopenia amegacariocítica 2 (MIM 616738 AD) |
Tabla 1. Se detectó una variante de nucleótido único (SNP) en el gen MECOM (NM_004991.4), indicando cigosidad, clasificación de patogenicidad y el fenotipo asociado según OMIM.
Posteriormente, ante la descripción de síndrome asociado al gen MECOM, se evaluó la posible asociación con sinostosis radiocubital y compromiso óseo. Con un examen físico sin anormalidades, incluyendo pronosupinación conservada. Se realizaron radiografías de miembros superiores y estudios óseos complementarios, sin identificarse alteraciones estructurales.
Finalmente, tras confirmar el diagnóstico de síndrome asociado a MECOM con insuficiencia medular severa, el paciente fue remitido al equipo de trasplante de médula ósea para evaluación y estudio como única opción terapéutica curativa.
Discusión
Los fallos medulares congénitos constituyen un grupo heterogéneo de enfermedades caracterizadas por insuficiencia medular en etapas tempranas de la vida, que conduce a citopenias de una o varias líneas celulares. En los últimos años, esto ha impulsado el estudio de variantes genéticas en diferentes genes, entre los cuales se ha identificado una amplia gama de alteraciones en las secuencias del locus MECOM(6).
El locus MECOM es un sitio genético complejo que codifica múltiples transcritos, implicados en la regulación hematopoyética y en procesos de oncogénesis. Se reconoce además como una región de alta inestabilidad, en la que diversas alteraciones genéticas pueden dar lugar a un síndrome heterogéneo y poco frecuente(6). La mayoría de las mutaciones descritas se localizan en la región que codifica los diez dominios de dedos de zinc del locus, considerados puntos de alta frecuencia mutacional(7).
Las características de las proteínas EVI1 y la isoforma MDS1–EVI1 fueron descritas por primera vez en 1991. Estas poseen dos grupos de dominios de dedos de zinc que les permite unirse al ADN: un grupo N-terminal, que abarca los exones 3 al 8 y contiene siete dedos de zinc con afinidad por secuencias específicas de ADN, y un grupo C-terminal, formado por los exones 9 al 13, con tres dedos de zinc que modulan la actividad transcripcional(7,8). En el caso descrito se identificó una variante en el exón 8, ubicada al final del grupo N-terminal, lo que interrumpe la síntesis proteica antes de completar los siete primeros dedos de zinc y elimina los tres del extremo C-terminal, resultando en pérdida total de la capacidad de unión al ADN y disfunción del factor de transcripción EVI1, lo que explica el fenotipo y compromiso hematológico grave observado.
En modelos animales, como ratones, se ha encontrado expresión de este gen en tejido pulmonar, urinario, cardiovascular y el esbozo de las extremidades durante la embriogénesis, lo que explica las diferentes presentaciones clínicas que este síndrome puede presentar y refleja la fisiopatología observada en humanos(9).
En la literatura se han reportado múltiples variantes asociadas a fenotipos distintos, en las que se incluye mutaciones sin sentido (nonsense), deleciones, mutaciones en sitios de empalme o de sentido erróneo (missense)(10). El fenotipo mayormente asociado a variante sin sentido ha sido la insuficiencia medular congénita asociada a sinostosis radiocubital. Así mismo, se han documentado casos de malformaciones cardíacas como: defecto del tabique auricular, cardiopatías cianosantes y foramen oval permeable asociado a deleciones o mutaciones en el sitio de empalme(10).
La principal alteración esquelética descrita es la sinostosis radiocubital, que puede presentarse con o sin trombocitopenia amegacariocítica (RUSAT). Este último se caracteriza por la fusión congénita proximal del radio y el cúbito, con limitación de la pronosupinación del antebrazo y trombocitopenia progresiva hacia pancitopenia(9). Estas manifestaciones se han asociado con mayor frecuencia a variantes sin sentido.
En 2023 se publicó un estudio que incluyó ocho pacientes no relacionados portadores de variantes en MECOM, entre las cuales se identificaron deleciones completas o parciales, mutaciones con sentido erróneo y alteraciones que afectaban los sitios de empalme. Además de las manifestaciones hematológicas, los pacientes presentaron compromiso óseo, incluyendo sinostosis radiocubital, clinodactilia e hipoplasia radial; anomalías cardíacas como ductus arterioso persistente y comunicaciones interauricular o interventricular; y alteraciones renales como hipoplasia y malrotación. También se observaron rasgos craneofaciales caracterizados por micrognatia y macro o microcefalia, así como retraso del desarrollo, hipoacusia, hipotonía y fallo del medro. Estos hallazgos evidencian la amplia variabilidad fenotípica asociada a las diferentes variantes patogénicas descritas en MECOM(11).
El primer caso reportado de síndrome asociado a MECOM con un fenotipo caracterizado por fallo medular, publicado en 2012, correspondiente a una paciente que debutó a los dos días de vida con petequias faciales y trombocitopenia grave, que progresó a pancitopenia a los dos meses. El análisis mediante microarreglo de SNP identificó una deleción de novo en 3q26.2, que abarcaba los genes EVI1, MDS1 y C3orf50, recibiendo trasplante alogénico de médula ósea a los 4 meses(12).
Posteriormente en 2018, se publicaron dos estudios que ampliaron la caracterización molecular de este síndrome. En el primero, se analizaron nueve pacientes con trombocitopenia congénita e insuficiencia medular sin causa aparente, identificando deleciones y mutaciones heterocigotas en MECOM. Se determinó que las mutaciones sin sentido y los cambios de marco de lectura provocaban terminación prematura de la traducción proteica, mientras que las mutaciones con sentido erróneo alteraban el dominio de unión al ADN de EVI1, afectando su función; las deleciones, por su parte, causaban haploinsuficiencia(13). El mismo año, se reportaron 12 pacientes con mutación en locus MECOM: siete debutaron con insuficiencia medular, dos con pancitopenia severa, detectando la mutación c.2248C>T (p.Arg750Trp), los 5 restantes, presentaron como manifestación inicial pancitopenia grave desde el nacimiento o trombocitopenia aislada con progresión a fallo medular durante las primeras semanas de vida(1).
De acuerdo con la evidencia disponible, los pacientes con insuficiencia medular congénita sin anomalías esqueléticas suelen presentar variantes sin sentido, de inserción/deleción o de empalme que generan un codón de terminación prematura en MECOM(10,11). En este contexto, el caso descrito representa la primera asociación clínica documentada con la variante MECOM c.2242 C>T (p.Arg 748*), la cual se relaciona con un fenotipo hematológico severo.
El reporte de exoma señalaba que las variantes patogénicas en heterocigosis en MECOM están asociadas con sinostosis radiocubital con trombocitopenia amegacariocítica 2. Sin embargo, en este paciente no se evidenció compromiso óseo, lo que refleja la heterogeneidad fenotípica de las alteraciones en este locus.
Este reporte, por tanto, amplía el espectro genotípico y fenotípico conocido de esta entidad, contribuyendo a una mejor comprensión de las correlaciones entre el tipo de alteración molecular y la variabilidad de la presentación clínica observada.
Conclusión
El conocimiento de este caso permite enfatizar que las mutaciones asociadas a MECOM deben considerarse dentro del diagnóstico diferencial de la pancitopenia severa del recién nacido y del lactante, representando una de las posibles causas genéticas de insuficiencia medular, incluso en ausencia de malformaciones esqueléticas asociadas. La identificación genética temprana permite definir la necesidad de trasplante de médula ósea precoz como única medida curativa y orientar el seguimiento multidisciplinario del paciente y su familia.
Declaración de conflictos de interés: los autores declaran no poseer conflictos.
Bibliografía
1. Germeshausen M, Ancliff P, Estrada J y col. MECOM-associated syndrome: a heterogeneous inherited bone marrow failure syndrome with amegakaryocytic thrombocytopenia. Blood Adv. 2018;2:586-596. https://doi.org/10.1182/bloodadvances.2018016501
2. Ammeti D, Marzollo A, Gabelli M y col. A novel mutation in MECOM affects MPL regulation in vitro and results in thrombocytopenia and bone marrow failure. Br J Haematol. 2023;203:852-859. https://doi.org/10.1111/bjh.19023
3. Steinleitner K, Rampetsreiter P, Köffel R y col. EVI1 and MDS1/EVI1 expression during primary human hematopoietic progenitor cell differentiation into various myeloid lineages. Anticancer Res. 2012;32:4883-4889.
4. Weizmann D, Pincez T, Roussy M y col. New MECOM variant in a child with severe neonatal cytopenias spontaneously resolving. Pediatr Blood Cancer. 2020;67:e28215. https://doi.org/10.1002/pbc.28215
5. Voit RA, Sankaran VG. MECOM Deficiency: from Bone Marrow Failure to Impaired B-Cell Development. J Clin Immunol. 2023;43:1052-1066. https://doi.org/10.1007/s10875-023-01545-0
6. Feurstein S. Emerging bone marrow failure syndromes- new pieces to an unsolved puzzle. Front Oncol. 2023;13:1128533. https://doi.org/10.3389/fonc.2023.1128533
7. Huang D, Jiang M, Zhu Y y col. A novel missense mutation in the MECOM gene in a Chinese boy with radioulnar synostosis with amegakaryocytic thrombocytopenia. BMC Pediatr. 2024;24:62. https://doi.org/10.1186/s12887-024-04552-1
8. Perkins AS, Fishel R, Jenkins NA, Copeland NG. Evi-1, a murine zinc finger proto-oncogene, encodes a sequence-specific DNA-binding protein. Mol Cell Biol. 1991;11:2665-2674. https://doi.org/10.1128/mcb.11.5.2665-2674.1991
9. Nagai K, Niihori T, Muto A y col. Mecom mutation related to radioulnar synostosis with amegakaryocytic thrombocytopenia reduces HSPCs in mice. Blood Adv. 2023;7:5409-5420. https://doi.org/10.1182/bloodadvances.2022008462
10. Li J, Peng T, Cheng G y col. A novel MECOM gene variant causes severe thrombocytopenia in a neonate: a case report and review of the literature. J Med Case Rep. 2025;19:147. https://doi.org/10.1186/s13256-025-05194-2
11. Lozano Chinga MM, Bertuch AA, Afify Z y col. Expanded phenotypic and hematologic abnormalities beyond bone marrow failure in MECOM-associated syndromes. Am J Med Genet A. 2023;191:1826-1835. https://doi.org/10.1002/ajmg.a.63208
12. Nielsen M, Vermont CL, Aten E y col. Deletion of the 3q26 region including the EVI1 and MDS1 genes in a neonate with congenital thrombocytopenia and subsequent aplastic anaemia. J Med Genet. 2012;49:598-600. https://doi.org/10.1136/jmedgenet-2012-100990
13. Kjeldsen E, Veigaard C, Aggerholm A, Hasle H. Congenital hypoplastic bone marrow failure associated with a de novo partial deletion of the MECOM gene at 3q26.2. Gene. 2018;656:86-94. https://doi.org/10.1016/j.gene.2018.02.061