Bone marrow changes in a case of Wilson’s disease- A Case report
Drishti Sachan, Nashwa Kamal Shroff, Komal Yadav, Rajeev Sen
Hematology department, SGT Medical college and research institute, Gurugram, India.
Drishti Sachan- 1st Author: Orcid id- 0009-0007-6041-3973
Nashwa Kamal Shroff Orcid id- 0009-0008-8452-646X
Komal Yadav Orcid id- 0009-0008-5556-0976
Rajeev Sen Orcid id- 0000-0001-7957-5613
Keywords: Wilson’s disease, bicytopenia, perinuclear vacuolization
ABSTRACT
Background-
Wilson’s disease is a rare autosomal recessive disorder of copper metabolism caused by mutation of ATP7B gene on chromosome 13 resulting in excess accumulation of free copper in the liver, brain and eyes.
Case presentation-
We describe the case of a twelve year old boy with Wilson’s disease who developed bicytopenia. Bone marrow aspirate and biopsy was subsequently performed and the findings were observed. Informed consent was taken from the patient before the procedure.
Conclusion-
Bone marrow aspirate and biopsy revealed drug induced suppression of bone marrow. Myeloid, erythroid and megakaryocyte series showed perinuclear vacuolization along with abnormal pigment deposition in myeloid precursors. Close monitoring of hematological parameters is required while the patient is on treatment.
INTRODUCTION
Wilson’s disease is a rare autosomal recessive disorder of copper metabolism caused by mutation of ATP7B gene on chromosome 13 resulting in a systemic overload of copper. It is also known as hepatolenticular degeneration (hepato- liver, lenticular- brain), where the copper is deposited in brain, liver, kidney, eyes etc. 1 1 in 50,000 are diagnosed with Wilson’s disease.2
The main complications of Wilson’s disease include brain damage and liver cirrhosis, psychiatric disturbances, i.e., depression, suicidal tendencies, aggressive behaviour motor dysfunction and corneal opacities.3
Pancytopenia is an unusual initial presentation of this disease and when present, it diverts the usual diagnostic algorithm, thus delaying the treatment.4
Here we describe case of a 12 year old boy with Wilsons Disease who initially presented with neuropsychiatric symptoms and bicytopenia.
CASE PRESENTATION
A 12 year old boy was presented to the paediatric department with major complaint of difficulty in movements of all 4 limbs since last 5-6 months.
The child was apparently well about 5-6 months back when parents suddenly noted that he started developing weakness in both hands, more prominently noticed while writing in school, which gradually progressed to difficulty in walking, weakness of lower limbs, abnormal smiling at inappropriate times, irritable behaviour, difficulty in swallowing as they noticed that the child has started to eat very slowly associated with decreased performance in school.
They took the child to multiple practitioners, but there was no relief of any symptoms. His psychological evaluation report showed his total score on VSMS (SQ=77%) which indicated age-inappropriate social maturity, self- management and independence in adaptive skills/ activities of daily living.
Investigations such as MRI brain was suggestive of abnormal signal alteration in bilateral basal ganglia, thalami, mid-brain and pons with small patchy signal alteration in bilateral middle frontal cortical regions, favouring Wilson’s disease. The boy had past history of dengue fever about 4 months before the onset of present condition. One of his elder siblings died 2.5 years back, at 13 years of age with jaundice but the cause was unknown.
Clinical examination revealed conscious and alert child with abnormal smiling, ataxia, dysarthria, dysphagia, intentional tremors and Kayser- Fleischer rings.
The child underwent certain investigations result of which revealed (hemoglobin 11 gm/dl), leucopenia (TLC 3030/cumm), thrombocytopenia (platelet count –1x 109/L). RBC count and hematocrit (31.3%) was also reduced. RBC indices and RDW was within normal limits (MCV- 86.0, MCH-28.2, MCHC- 32.9, RDW- 14.4). Peripheral blood smear examination revealed normocytic normochromic RBCs, leucopenia and thrombocytopenia (bicytopenia). Prothrombin time was 13.60 sec and activated partial thromboplastin time was raised (37.10 sec). Serum iron profile was normal. Liver function tests showed normal bilirubin with marked increase in alkaline phosphatase (311 U/l) and other relevant parameters were within normal range. Serum lipase 451 U/L and serum amylase 127 U/L were elevated. Serum ceruloplasmin was <9.58 mg/dl (normal 18-50 mg/dl) and 24 hr urinary copper report was 107.91 microgram/day (normal 3-50 ug/day).
Ultrasound of the abdomen showed heterogeneously hypoechoic echotexture of liver with raised periportal echogenicity and splenomegaly. Patient was diagnosed as Wilsons disease and started on penicillamine therapy. Even after this therapy, the condition did not improve. Post therapy investigations revealed further leucopenia -2580/cumm (earlier 3030/cumm) and thrombocytopenia, 75,000/ μL (earlier 1x 109/L). The child underwent bone marrow aspiration and bone marrow biopsy for a detailed examination.
Bone marrow aspirate showed 30 % cellularity, which was hypocellular for this age. M:E ratio was 1.5:1 and myelogram revealed 30 % erythroid series, 45 % myeloid cells, normal number of lymphocytes and other cells. Erythroid and myeloid series revealed chromatin clumping, pyknotic nuclei and perinuclear vacuolization. Abnormal granule deposition is also noted in some cells of myeloid series. Megakaryocytes were normal in number and perinuclear vacuolization was noted. Iron stain was grade-4 with presence of large granules of iron in small clumps within the bone marrow particles. Bone marrow biopsy showed similar findings and reticulin was grade-2. Both aspirate and biopsy revealed drug induced toxic suppression of bone marrow. (Figures -1-5)
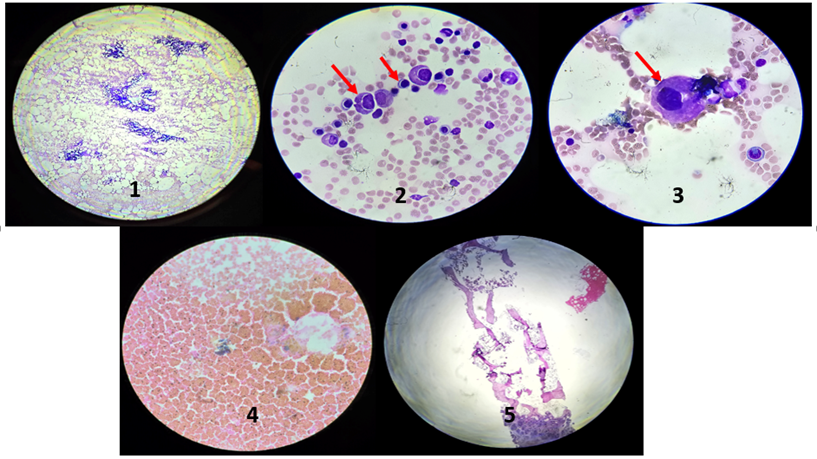
Figure 1- Bone marrow aspirate (scanner view-4x), Figure 2- Bone marrow aspirate showing perinuclear vacuolization in myeloid and erythroid series, 40x [red arrow], Figure 3- Bone marrow aspirate showing perinuclear vacuolization in megakaryocyte,40x [red arrow],Figure 4- Iron stain- Grade 4, Figure 5- Bone marrow biopsy showing hypocellular marrow for age.
DISCUSSION
In Wilson’s disease, serum copper and serum ceruloplasmin are less than normal and in response to penicillamine treatment, urinary copper excretion is initially markedly increased. Copper deficiency plays a significant role in the developement of bone marrow cytopenias in patients with Wilson’s disease, especially as a consequence of overtreatment with copper chelation therapy or excessive zinc supplementation. In Wilson’s disease, the standard treatment involves copper chelators (such as penicillamine) and/or zinc, which reduce copper accumulation. However, excessive chelation or high-dose zinc can cause copper deficiency (hypocupremia).5,6 Copper is a crucial cofactor for enzymes involved in hematopoiesis, including cytochrome oxidase and ceruloplasmin ferroxidase, which are essential for iron metabolism and erythropoiesis.7 Due to copper deficiency in Wilson’s disease patients, bone marrow changes such as vacuolization of erythroid and myeloid precursors and sometimes ringed sideroblasts is observed5. Patients on chelation therapy who develop unexplained cytopenias often show bone marrow dysplasia due to copper deficiency. Hematological abnormalities (anemia, neutropenia) improve with copper supplementation and/or cessation of excessive zinc or chelation therapy. In Wilson’s disease, serum copper and ceruloplasmin are low, so diagnosing copper deficiency relies on clinical context, bone marrow findings, and response to copper supplementation rather than serum copper levels alone. Repeated checking of blood counts in WD patients on chelation or zinc therapy is recommended to detect early signs of cytopenias, which may indicate impending copper deficiency.
In conclusion, copper deficiency is a recognized, reversible cause of bone marrow cytopenias in wilsons disease, particularly in the context of overtreatment. Regular monitoring of hematological parameters is crucial in patients undergoing chelation therapy to prevent and promptly address drug induced complications.
TABLE
|
Factor |
Impact on Bone Marrow |
Clinical Manifestation |
Reversibility |
|
Copper deficiency |
Trilineage dysplasia, vacuolization |
Anemia, neutropenia, ± thrombocytopenia |
Yes, with copper repletion |
|
Copper overload |
Hemolysis, membrane damage |
Hemolytic anemia, thrombocytopenia |
No (toxicity) |
Declaración de conflictos de interés: los autores declaran no poseer conflictos.
REFERENCES
(1) Shanmugam S, Kennady JA, Ahamed J. A case study on wilson’s disease. Int J Med Rev Case Rep. 2019; 3(3): 120-122. doi:10.5455/IJMRCR.wilson-disease
(2) Chanpong A, Dhawan A. Wilson disease in children and young adults - State of the art. Saudi J Gastroenterol. 2022; 28(1):21-31. doi: 10.4103/sjg.sjg_501_21.
(3) Bini V, Aloysius J, Syama Prasad TV, Reshma R, Remya R. A case report on wilson's disease-induced liver cirrhosis. Asian J Pharm Clin Res. 2017;10:113-4. doi:10.22159/ajpcr.2017.v10i4.16239
(4) Kaetz HW, Brodoff SS, Robbins AW. Wilson's Disease presenting as pancytopenia: splenectomy in management. Connecticut Medicine. 1966; 30(5):338-40.
(5) Rau AR, Usha M, Mallya P, Rau AT. Cytopenia and Bone Marrow Dysplasia in a Case of Wilson's Disease. Indian J Hematol Blood Transfus. 2014; 1:433-6. doi: 10.1007/s12288-014-0456-3.
6 ) Sharma S, Singh A, Sing P et al. Aplastic anemia in a patient with Wilson’s disease. Journal of hematology, North America, 2012; 1: 4-5. doi: https://doi.org/10.4021/jh39e
(7)Litwin T, Antos A, Bembenek J, et al. Copper Deficiency as Wilson's Disease Overtreatment: A Systematic Review. Diagnostics (Basel). 2023; 13(14):2424. doi: : https://doi.org/10.3390/diagnostics13142424.