Heterogeneidad molecular en mieloma múltiple
Molecular heterogeneity in multiple myeloma
Jorge López Villegas
Servicio de Hematología, Hospital Dr. Rafael Ángel Calderón Guardia, Caja Costarricense de Seguro Social, San José, Costa Rica.
jolovi85@hotmail.com / jlopezv@ccss.sa.cr
Identificador ORCID: https://orcid.org/0009-0004-1909-7065
Palabras clave: mieloma múltiple,
secuenciación de nueva generación,
mutaciones,
evolución clonal,
medicina de precisión,
estadificación de neoplasias.
Key words: multiple myeloma,
next generation sequencing,
mutations,
clonal evolution,
precision medicine,
neoplasm staging.
Glosario de abreviaturas:
ADN: ácido desoxirribonucleico
ARN: ácido ribonucleico
BCMA: B cell maturation antigen
CAR-T: chimeric antigen receptor T cells
EMR: enfermedad medible residual
GMSI: gammapatía monoclonal de significado incierto
MM: mieloma múltiple
DHMM: double hit multiple myeloma
MMND: mieloma múltiple de nuevo diagnóstico
MMRR: mieloma múltiple en recaída o refractario
MMS: mieloma múltiple smoldering
R-ISS: Revised International Staging System
SCS: single cell sequencing
scRNA-seq: single-cell ribonucleic acid sequencing
WES: whole exome sequencing
WGS: whole genome sequencing
Resumen
El mieloma múltiple es una enfermedad neoplásica de las células plasmáticas que representa la segunda neoplasia hematológica más común a nivel mundial. En la última década se han producido grandes avances en la caracterización molecular de esta enfermedad con la incorporación de nuevas tecnologías como secuenciamiento de nueva generación y secuenciamiento de célula única. El grado de heterogeneidad molecular intra e interpaciente observado en mieloma múltiple es mayor que en otros tipos de cáncer hematológicos, con cientos de mutaciones en decenas de genes distintos ocasionadas por diversos mecanismos. Además, se ha reportado la presencia de múltiples subclones neoplásicos en un mismo paciente. Estos subclones modifican su frecuencia en función de la etapa de la enfermedad, terapia y el sitio anatómico, lo cual tiene incidencia directa sobre la estratificación pronóstica y el tratamiento. Actualmente, la heterogeneidad molecular es el factor más relevante para establecer clasificaciones de riesgo y diseñar nuevas estrategias terapéuticas personalizadas en esta enfermedad.
Abstract
Multiple myeloma is a neoplastic disease of plasma cells that represents the second most common hematological neoplasm worldwide. In the last decade, great progress has been made in the molecular characterization of this disease with the incorporation of new technologies like next generation sequencing and single cell sequencing. The degree of intra and interpatient molecular heterogeneity observed in multiple myeloma is higher than that of other types of hematological cancers with hundreds of mutations in tenths of different genes generated by diverse mechanisms. Furthermore, the presence of multiple neoplastic subclones within the same patient has been reported. This subclones modify their frequency according to disease stage, therapy and anatomical site, which has a direct incidence over the prognostic stratification and treatment. Currently, molecular heterogeneity is the most relevant factor for establishing risk classifications and for designing new personalized therapeutic strategies in this disease.
Introducción
El mieloma múltiple (MM) es una neoplasia hematológica derivada de la proliferación descontrolada de células plasmáticas en la médula ósea. Es la segunda malignidad hematológica más frecuente a nivel mundial, representa anualmente el 10% de los casos de neoplasias hematológicas y el 1-2% si se consideran todos los tipos de cáncer(1,2).
En la última década se ha adquirido gran cantidad de información con respecto a la patogénesis del MM y su evolución. Mediante estudios de expresión génica y más recientemente de single cell sequencing (SCS) se ha podido determinar que el MM es una neoplasia con una alta heterogeneidad intra e interpaciente dada por la variabilidad en las alteraciones genéticas presentes en los distintos clones y subclones(3-5). A pesar de avances recientes en terapia y seguimiento, esta heterogeneidad es un obstáculo importante para mejorar la supervivencia de los pacientes. Con la aparición de nuevas técnicas moleculares nuestro entendimiento de las bases moleculares del MM ha crecido sustancialmente. Sin embargo, también ha puesto de manifiesto la complejidad genética de esta enfermedad. Comprender la heterogeneidad molecular del MM es clave para brindar una mejor atención a los pacientes y brindar en un futuro la alternativa de terapias individualizadas.
Esta revisión pretende sintetizar la literatura científica acerca de esta heterogeneidad molecular, así como su impacto sobre el pronóstico y perspectivas de tratamiento personalizado en los pacientes con MM.
Mecanismos de inestabilidad genética en células plasmáticas
Se ha postulado que alteraciones en el proceso de maduración de los linfocitos B, principalmente en los reordenamientos V(D)J y la RCC, son responsables de las traslocaciones observadas en MM. El hecho de que es frecuente observar mutaciones en genes asociados con la regulación de los eventos de modificación de IgH como IRF4, NDS2 y DIS3 en pacientes con mieloma múltiple refuerza esta teoría. Además, se han descrito firmas mutacionales asociadas con actividad aumentada de las enzimas involucradas en la mutación hipersomática y la recombinación de cambio de clase, como las de la familia APOBEC y AID con presencia de mutaciones en cluster, inestabilidad genómica incrementada y peor pronóstico(6,7). Se propone que desregulación en AID contribuye a la adquisición de mutaciones en etapas tempranas mientras que firmas mutacionales asociadas a APOBEC se correlacionan con enfermedad de alto riesgo y progresión(8).
En MM también se ha descrito la presencia de reordenamientos complejos del genoma derivados de eventos de cromoplexia, inserciones templadas y cromotripsis en un alto porcentaje de los pacientes, a diferencia de lo que ocurre en otras neoplasias hematológicas(6,9,10).
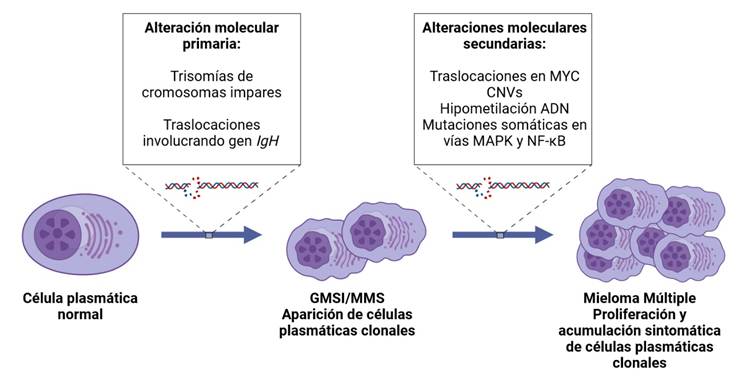
Figura 1. Eventos moleculares desencadenantes en MM(2,11).
Eventos moleculares primarios y secundarios en MM
Se propone que la aparición de gammapatía monoclonal de significado incierto (GMSI) y mieloma múltiple tipo smoldering (MMS), estados preneoplásicos que preceden al MM en la gran mayoría de los pacientes, ocurren por un evento primario asociado con hiperdiploidía dada por trisomías en cromosomas impares aproximadamente en el 40-60% de los casos y traslocaciones del cromosoma 14 que involucran al gen IGH en el resto de los casos, con una cantidad baja de pacientes que pueden presentar ambas(1,2,11,12).
En el caso de los MM con translocación en IGH, el cambio estructural genera que oncogenes como CCND1, CCND3, MAF, MAFB, NSD2 y FGFR3 queden bajo el control del potenciador de IGH, aumentando significativamente su expresión y generando desregulación en el complejo de control celular de las ciclinas, específicamente en el punto de control G1/S(2,13,14). La hiperdiploidía también se ha asociado con afectación del punto de control G1/S, pero los mecanismos de adquisición de las alteraciones y cómo las trisomías generan este efecto aún no se ha dilucidado, aunque se postula que deriva de la sobreexpresión de genes que promueven replicación desregulada.
Posterior a este evento primario (traslocación IGH o hiperdiploidía), las células plasmáticas sufren eventos secundarios relacionados con adquisición de ventajas proliferativas y desregulación del ciclo celular, como alteraciones del gen MYC en el locus 8q24, deleción 17p, mutación de TP53, ganancia o amplificación de 1q, cambios de hipometilación del ácido desoxirribonucleico (ADN) y mutaciones en genes de las vías de señalización RAS/MAPK y NF-κB(2,6,13).
Técnicas moleculares para el abordaje de heterogeneidad en MM
La técnica de next generation sequencing (NGS) ha sido clave para descifrar la evolución clonal desde las células preneoplásicas de la GMSI y el MMS hasta las etapas avanzadas de la enfermedad. Desde el 2012, Keats y colaboradores analizaron muestras de 28 pacientes en distintas etapas clínicas mediante microarreglos de hibridación genómica comparativa y generaron, además, un modelo de competencia clonal en ratón, que evaluaron con secuenciación de genoma completo(15). Los autores describieron 3 modalidades de evolución en MM: genéticamente estable en pacientes con riesgo estándar y evolución linear y evolución ramificada con mezclas de subclones que alternan en dominancia a través del tiempo en pacientes con alteraciones citogenéticas de alto riesgo. Simultáneamente, Egan y colaboradores llegaron a una conclusión similar evaluando por secuenciación de genoma completo muestras seriadas de un paciente con MM y t(4;14) durante un período de 5 años(13).
Posteriormente, Rasche y colaboradores publicaron un estudio que integraba estos conceptos evolutivos con la primera descripción de heterogeneidad espacial en MM, añadiendo un grado de complejidad mayor al tema. Los autores usaron secuenciación de exoma completo en 42 muestras de pacientes con MMND de diferentes sitios anatómicos y observaron que las células neoplásicas seguían un modelo de evolución Darwiniano con predominio de clones más aptos para colonizar nichos en médula ósea, que muchas veces desplazan completamente a las células normales, y otros clones en oleadas, lo que se conoce como “clonal sweeps”. Después de estos eventos, las células neoplásicas evolucionan de forma regional con múltiples subclones que se generan en sitios distintos y acumulan alteraciones moleculares diferenciadas(16).
En cuanto al valor pronóstico de las mutaciones detectadas por NGS se ha observado que éste es menor que el de las alteraciones cromosómicas. Sin embargo, se ha logrado evidenciar un efecto aditivo de las mutaciones driver sobre la agresividad de la enfermedad, dando mayor preponderancia a la carga mutacional observada que a mutaciones específicas (con excepción de mutaciones en algunos genes como TP53 y ZFHX4)(17-19). El reconocimiento de pacientes de alto riesgo que no son identificados por el R-ISS (DHMM) ha puesto de manifiesto la necesidad de incorporar información molecular adicional dentro de los modelos de estadificación, y NGS es una técnica que en el futuro permitirá obtener esta información de forma confiable, rápida y costo efectiva(19).
La posibilidad de estudiar de forma individual células tumorales de MM y su microambiente ha abierto una nueva área de investigación en esta enfermedad. Las técnicas de secuenciación convencionales trabajan a partir de agregados de material genético por lo cual no son capaces de abarcar de la mejor forma la alta heterogeneidad molecular del MM. Las técnicas de SSC permiten un estudio detallado de las alteraciones moleculares y niveles de expresión génica a nivel subclonal, por lo que son muy valiosas para determinar la estructura clonal de la neoplasia de un paciente y dar seguimiento a los cambios generados por la terapia en la frecuencia y composición genómica de dichos clones.
Ledergor y colaboradores fueron los primeros investigadores en aplicar esta nueva tecnología para la obtención de perfiles de expresión de ácido ribonucleico a partir de células individuales mediante single cell ribonucleic acid sequencing (scRNA-seq) de 29 pacientes con estados preneoplásicos y mieloma múltiple de nuevo diagnóstico (MMND), así como 11 controles normales. Los autores identificaron que cada paciente presentaba un perfil transcripcional distinto. Sin embargo, algunos compartían características comunes, como la sobreexpresión de genes asociados al control del ciclo celular (CCND1 y CCND2) y a la regulación de la transcripción (FGFR3). Un hallazgo interesante de este estudio fue que el grado de heterogeneidad en los perfiles que presentaron las células neoplásicas de los pacientes analizados no dependía directamente del perfil mutacional de los genes, lo que indica que el análisis de alteraciones en regiones no codificantes, modificaciones epigenéticas y ambientales pueden ser especialmente relevantes en MM para la diversidad clonal y subclonal.
Otro ámbito en el cual las técnicas de SSC han sido muy útiles es en el estudio de la heterogeneidad espacial del MM. Esta enfermedad se caracteriza por tener una distribución irregular en la médula ósea, frecuentemente organizándose en zonas de crecimiento que semejan tumores sólidos, en lo que se conoce como lesiones focales, cuyo tamaño y número aumentado se ha asociado con mal pronóstico(16,20,21). Además, en algunos pacientes en estados avanzados se pueden generar lesiones óseas paramedulares o inclusive leucemia de células plasmáticas. John y colaboradores analizaron 16 muestras pareadas (médula ósea/lesión focal) de pacientes con MMND mediante whole genome sequencing (WGS) y scRNA-seq y encontraron diferencias significativas a nivel cromosómico y/o mutacional en 12 de ellos(21). En estos 12 pacientes identificaron clones que se presentaban exclusivamente en las lesiones focales, con mayor número de variantes de un solo nucleótido en genes driver. En total, los investigadores detectaron un promedio de 6 subclones por paciente, algunos de los cuales no estaban presentes en la médula ósea del todo o representaban subclones minoritarios(21).
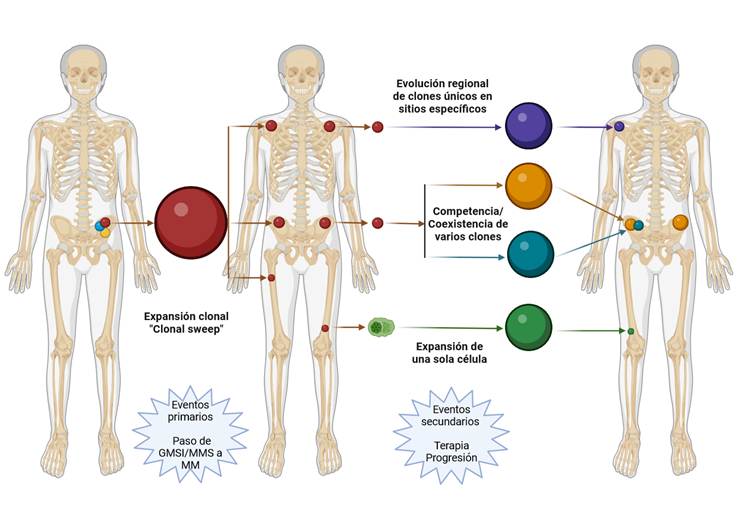
Figura 2. Patrones de evolución clonal en MM. (15,16,21–23)
El grupo de Rasche y colaboradores dio seguimiento a los estudios que habían realizado previamente en heterogeneidad espacial(16), analizando 140 muestras de médula ósea y lesiones focales en 24 pacientes con MM obtenidas durante un período de hasta 14 años. Los autores observaron que el mayor grado de heterogeneidad ocurre en las lesiones focales, mientras que el infiltrado difuso en médula ósea tiende a ser mucho más homogéneo en términos de número y tipo de alteraciones moleculares(22). Además, los investigadores describen 3 patrones evolutivos, similares a los propuestos por Keats et al en 2012: expansión de una sola célula, evolución de clones en competencia/coexistentes y clones únicos sitio específicos (ver Figura 2)(15,22). Previamente Landau y colaboradores también habían reportado la expansión clonal acelerada a partir de una sola célula posterior a tratamiento con melfalán y trasplante autólogo(23). Cabe destacar que el modelo propuesto puede tener implicancias importantes desde el punto de vista terapéutico, ya que el clon no sería detectado en análisis basales de whole exome sequencing (WES) o WGS a partir de ADN bulk.
Impacto de la heterogeneidad molecular de MM sobre pronóstico y tratamiento
El avance en técnicas moleculares en años recientes ha demostrado que los sistemas de clasificación de riesgo en MM basados únicamente en características clínicas y/o análisis citogenético convencional no permiten identificar adecuadamente grupos de pacientes con riesgo alto, lo cual ha obligado a reformular estas herramientas(1,24). En el caso del International Staging System (ISS), en años recientes ha sido actualizado en dos oportunidades, en el 2016 para añadir t(4;14), t(14;16) y del(17p) como factores de alto riesgo en la primera revisión (R-ISS) y posteriormente, en 2022, para incluir la ganancia/amplificación de 1q en la segunda revisión (R2-ISS). Estas modificaciones han permitido clasificar de mejor manera pacientes con R-ISS II, reasignándoles características de riesgo intermedio-bajo o intermedio-alto(1,20,25). Además, el reconocimiento de una nueva categoría de riesgo muy alto denominada Double Hit Multiple Myeloma (DHMM), la cual involucra la inactivación bialélica de TP53 o amplificación (≥4 copias) de CKS1B en el contexto de una clasificación grupo III bajo los criterios del ISS, ha sido muy relevante, identificando pacientes con un pronóstico especialmente adverso en los cuales el uso de citogenética convencional no es suficiente para detectar las alteraciones relacionadas(24). Sin embargo, ninguno de los sistemas de clasificación actuales toma en cuenta la coocurrencia e interacción de diferentes alteraciones que ha sido demostrada por distintos estudios(6). Es probable que en los próximos años, la información que están generando las técnicas de secuenciación masiva bulk y single cell sea incorporada de alguna forma en la estratificación de los pacientes(26).
Por otra parte, la heterogeneidad molecular intra e interpaciente es el principal reto para un tratamiento óptimo de los pacientes que permita alcanzar tasas de remisión y supervivencia similares a las observadas en otras neoplasias hematológicas. Especialmente importante es la variabilidad clonal en distintos sitios anatómicos de un mismo paciente(27). El patrón de evolución ramificado y la heterogeneidad espacial que se presenta en muchos casos de MM al momento de la presentación y post tratamiento dificulta la erradicación de todos los clones y subclones presentes, e inclusive hay evidencia de preexistencia de clones resistentes a la terapia previo a la exposición a agentes terapéuticos(5).
Un aspecto relevante que añade complejidad es la heterogeneidad molecular aportada por las células neoplásicas presentes en lesiones osteolíticas extramedulares. Usando scRNA-seq y WGS, Merz et al estudiaron las diferencias espaciales genómicas entre células plasmáticas patológicas provenientes de médula ósea y células plasmáticas patológicas de lesiones osteolíticas. Los autores encontraron diferencias en el perfil de expresión génica de ambas poblaciones, observando un aumento en la expresión en las células plasmáticas de lesiones osteolíticas de genes asociados con enfermedad ósea y progresión en MM tales como TIMP1, HGF y LAMP5, así como, regulación negativa de JUN/FOS, DUSP1 y HBB(3). En un estudio similar, pero analizando lesiones de mayor tamaño (>1 cm de diámetro), John y colaboradores observaron mayores diferencias espaciales entre células plasmáticas que las descritas por el grupo de Merz(3,21). De forma interesante, clones genéticamente idénticos mostraron diferencias a nivel transcripcional y epigenético, principalmente en genes que codifican para componentes del complejo mayor de histocompatibilidad y blancos de inmunoterapia, lo cual puede tener implicancias terapéuticas(21).
Ante este panorama, los esfuerzos se han centrado sobre vías comunes afectadas en la mayoría de los pacientes, como es el caso de la vía ERK/MAPK(28). Se han realizado ensayos clínicos de fase 1 y fase 2 con resultados mixtos usando inhibidores de BRAF como vemurafenib e inhibidores de MEK como trametinib como agentes únicos en pacientes con MM en recaída/refractario (MMRR), con obtención de respuestas parciales pero que no perduran(5,29). Recientemente, Giesen y colaboradores reportaron los resultados obtenidos en el estudio GMMG-BIRMA usando una combinación de inhibidores de BRAF y MEK (encorafenib y binimetinib) en pacientes con MMRR y mutación BRAFV600E. Se observó una tasa de respuesta global de 83.3%, mejorando el desempeño de estudios previos que evaluaron monoterapia. Sin embargo, la duración de la respuesta fue baja (4.8 meses) y se identificó aparición de resistencia por expansión de subclones con mutaciones en NRAS (30). Ensayos clínicos de fase 1 y fase 2 analizando el bloqueo de otras vías de señalización, como PI3K/AKT, o blancos alternativos innovadores, como las quinasas tipo PIM (de proviral insertion site of Moloney murine leukemia virus) o c-MYC, no han generado resultados esperanzadores, con bajas tasas de respuesta(29).
Un factor común que explica la baja efectividad de las estrategias antes mencionadas es la alta variabilidad molecular de los clones y subclones presentes en sitios anatómicos distintos. Idealmente, el blanco de terapias dirigidas en MM deberían ser los eventos primarios, los cuales son compartidos por todos los subclones que se generan después de la transformación neoplásica, tales como las alteraciones en NDS2 y FGFR3 en pacientes con t(4;14). Sin embargo, estrategias contra estos eventos primarios aún son conceptuales y se encuentran lejos de la aplicación clínica(27,29).
Adicionalmente a las innovaciones farmacológicas, se ha tratado de mejorar el manejo de los pacientes con MM a través de estrategias que permitan considerar la heterogeneidad molecular para definir los esquemas de tratamiento. Por ejemplo, el uso de técnicas de imágenes funcionales, como la tomografía por emisión de positrones en combinación con secuenciación de muestras de lesiones focales, ha permitido establecer una relación entre el tamaño y número de las lesiones con el grado de heterogeneidad molecular presente en un paciente determinado(5). De esta forma, la caracterización de las lesiones focales puede convertirse en un futuro cercano en un marcador sencillo que permita identificar a estos pacientes con mayor heterogeneidad y riesgo de recaída/refractariedad, sin necesidad de secuenciar muestras de múltiples sitios anatómicos. En los últimos años, la enfermedad medible residual (EMR) por técnicas moleculares o citometría de flujo de nueva generación a niveles de sensibilidad de 10-5/10-6 ha demostrado impacto pronóstico en pacientes con MM, con resultados negativos asociados a mejor PFS y OS, independientemente del esquema de tratamiento o de la categoría de riesgo asignada al diagnóstico(31). Sin embargo, al realizar la EMR se analiza únicamente un aspirado de cresta iliaca que, debido a la alta heterogeneidad molecular y a la presencia de lesiones focales que se mantienen restringidas a ciertos sitios, en la mayoría de los casos no es representativo del estado real del paciente. Ante esto, es necesario obtener una perspectiva más amplia mediante la combinación de técnicas de imágenes funcionales, EMR y biopsia líquida(5). El uso de biopsia líquida para evaluar respuesta a tratamiento y probabilidad de recaída es especialmente prometedor, ya que permitiría identificar, caracterizar y dar seguimiento a subclones circulantes de distintos sitios anatómicos que pueden no ser detectados en un aspirado de cresta ilíaca. Por ahora los resultados obtenidos en diversos estudios que evalúan el uso de biopsia líquida para EMR son mixtos, pero el uso de técnicas más sensibles y mejores herramientas de procesamiento bioinformático pueden cambiar el panorama rápidamente(5,31).
Frente a las dificultades terapéuticas que presenta el MM y el gran porcentaje de pacientes que recaen y se vuelven refractarios a fármacos convencionales, la aplicación de tratamientos basados en principios inmunes ha representado un gran avance en los últimos años. La ventaja de estos tratamientos es que, al no depender directamente de las complejas características moleculares de las células neoplásicas, reducen el impacto de la heterogeneidad intra e interpaciente. El principal representante de este nuevo tipo de terapias en MM es el anticuerpo monoclonal anti CD38 daratumumab aprobado en 2016 por la Agencia Europea de Medicamentos (EMA: European Medicines Agency)(5). El daratumumab ha sido ampliamente incorporado en los esquemas de tratamiento más recientes con muy buenos resultados(20). Lamentablemente, ha probado ser ineficaz en pacientes con MM extramedular, lo cual ha generado cuestionamientos con respecto a su utilidad en pacientes con heterogeneidad espacial(5). Además, Vo y colaboradores han descrito recientemente la aparición de mutaciones en respuesta al tratamiento con daratumumab que resultan en la pérdida del epítopo al cual va dirigido el anticuerpo(28). Otra terapia novedosa evaluada en MM que tiene el potencial de sobreponerse a la heterogeneidad molecular es el uso de células T con receptor de antígeno quimérico (CAR-T: Chimeric Antigen Receptor T cells). En múltiples estudios clínicos, las células CAR-T dirigidas contra el antígeno de maduración de células B (BCMA: B Cell Maturation Antigen) han demostrado tasas de respuesta superiores al 80% y se han posicionado como una opción valiosa en pacientes con MMRR(32,33).
En un futuro cercano la caracterización molecular y fenotípica de las células plasmáticas tumorales en pacientes con MMND podría servir no únicamente para predicción del riesgo sino también para la selección de los agentes terapéuticos más efectivos contra los clones/subclones del paciente. En este sentido, los ensayos ex-vivo podrían representar un avance importante en terapia personalizada en MM(26). Kropivsek y colaboradores analizaron muestras de médula ósea de 70 pacientes en diferentes etapas de la enfermedad con técnicas de microscopía automatizada e inmunofluorescencia analizadas con inteligencia artificial, scRNA-seq y espectrometría de masas, y lo correlacionaron con las características clínicas. Los autores lograron identificar 4 perfiles fenotípicos distintos que tenían diferentes respuestas a fármacos e inmunoterapia ex-vivo y que eran predictivos de la respuesta de los pacientes a los esquemas de tratamiento elegidos por los médicos tratantes(34). Por ejemplo, identificaron un grupo de pacientes en etapas avanzadas con un ambiente proinflamatorio con aumento de factor de necrosis tumoral alfa (TNF-α) e interleukina 6 (IL-6) y alta infiltración por monocitos y células T que, paradójicamente, tenían una respuesta pobre a inmunoterapia en distintas combinaciones, tanto ex-vivo como clínica(34). De forma interesante, los autores proponen que sus hallazgos podrían abrir la posibilidad de integrar este tipo de información a la rutina clínica mediante técnicas como citometría de flujo, que permitan caracterizar la composición celular de la muestra a un nivel más macro mediante paneles de monitoreo inmune que permitan elegir el tratamiento con mayor probabilidad de éxito en un grupo de pacientes determinado tanto en MMND como en MMRR(34).
En Latinoamérica la disponibilidad de técnicas de SSC y transcriptómica espacial que permitirían obtener la información necesaria para un tratamiento personalizado ajustado a la heterogeneidad molecular de las células neoplásicas de cada paciente sigue siendo limitada a centros altamente especializados. En países con niveles de renta bajos o medios como los de nuestra región, existen varios obstáculos que dificultan la implementación de estas técnicas. La ausencia de infraestructura y equipamiento adecuados, la reticencia para conformar grupos de trabajo multidisciplinarios y de colaboración internacional y la dificultad de obtener financiamiento estatal o privado para proyectos de investigación relacionados con este tema son las principales barreras para la generación de datos relacionados con heterogeneidad molecular en mieloma múltiple en Latinoamérica. Afortunadamente, existen señales esperanzadoras, como el avance acelerado de las estrategias bioinformáticas para el análisis de los datos obtenidos por SSC, las cuales suelen ser de acceso libre(35). Además, cada vez son más frecuentes las alianzas público-privadas entre organismos gubernamentales y empresas farmacéuticas o biotecnológicas que permiten obtener recursos para inversión en nuevas tecnologías.
Conclusión
El MM presenta una gran diversidad genómica que la diferencia de otras neoplasias hematológicas donde la carga mutacional es mucho más baja. El advenimiento de las técnicas de NGS y SCS han permitido alcanzar altos niveles de detalle en la caracterización molecular de los distintos clones y subclones neoplásicos y seguirán aportando información valiosa con miras a incorporarlas, en un futuro cercano, a la práctica clínica como un primer paso hacia una terapia verdaderamente personalizada.
Conflicto de intereses. Ninguno para declarar.
Bibliografía
1. Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97:1086-107. DOI: 10.1002/ajh.26590.
2. Awada H, Thapa B, Awada H y col. A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells. 2021;10:1961. DOI: 10.3390/cells10081961.
3. Merz M, Merz AMA, Wang J y col. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat Commun. 2022;13:807. DOI: 10.1038/s41467-022-28266-z.
4. Walker BA, Wardell CP, Melchor L y col. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120:1077-86. DOI: 10.1182/blood-2012-03-412981.
5. Rasche L, Kortüm K, Raab M, Weinhold N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. Int J Mol Sci. 2019;20:1248. DOI: 10.3390/ijms20051248.
6. Aksenova AY, Zhuk AS, Lada AG y col. Genome Instability in Multiple Myeloma: Facts and Factors. Cancers. 2021;13:5949. DOI: 10.3390/cancers13235949.
7. Ashby C, Boyle EM, Bauer MA, y col. Structural variants shape the genomic landscape and clinical outcome of multiple myeloma. Blood Cancer J. 2022;12:85. DOI: 10.1038/s41408-022-00673-x.
8. Neuse CJ, Lomas OC, Schliemann C y col. Genome instability in multiple myeloma. Leukemia. 2020;34:2887-97. DOI: 10.1038/s41375-020-0921-y.
9. Maura F, Boyle EM, Rustad EH y col. Chromothripsis as a pathogenic driver of multiple myeloma. Semin Cell Dev Biol. 2022;123:115-23. DOI: 10.1016/j.semcdb.2021.04.014.
10. Maura F, Bolli N, Angelopoulos N y col. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun. 2019;10:3835. DOI: 10.1038/s41467-019-11680-1.
11. Solimando AG, Da Vià MC, Cicco S y col. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J Clin Med. 2019;8:997. DOI: 10.3390/jcm8070997.
12. Saxe D, Seo E, Beaulieu Bergeron M, Han J. Recent advances in cytogenetic characterization of multiple myeloma. Int J Lab Hematol. 2019;41:5-14. DOI: 10.1111/ijlh.12882.
13. Egan JB, Shi CX, Tembe W y col. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060-6. DOI: 10.1182/blood-2012-01-405977.
14. Pawlyn C, Davies FE. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood. 2019;133:660-75. DOI: 10.1182/blood-2018-09-825331.
15. Keats JJ, Chesi M, Egan JB y col. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067-76. DOI: 10.1182/blood-2012-01-405985.
16. Rasche L, Chavan SS, Stephens OW y col. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8:268. DOI: 10.1038/s41467-017-00296-y.
17. Bolli N, Biancon G, Moarii M y col. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604-16. DOI: 10.1038/s41375-018-0037-9.
18. Hu Y, Chen W, Wang J. Progress in the identification of gene mutations involved in multiple myeloma. OncoTargets Ther. 2019;12:4075-80. DOI: 10.2147/OTT.S205922.
19. Bolli N, Genuardi E, Ziccheddu B, Martello M, Oliva S, Terragna C. Next-Generation Sequencing for Clinical Management of Multiple Myeloma: Ready for Prime Time? Front Oncol. 2020;10:189. DOI: 10.3389/fonc.2020.00189.
20. Zamagni E, Barbato S, Cavo M. How I treat high-risk multiple myeloma. Blood. 2022;139:2889-903. DOI: 10.1182/blood.2020008733.
21. John L, Poos AM, Brobeil A y col. Resolving the spatial architecture of myeloma and its microenvironment at the single-cell level. Nat Commun. 2023;14:5011. DOI: 10.1038/s41467-023-40584-4.
22. Rasche L, Schinke C, Maura F y col. The spatio-temporal evolution of multiple myeloma from baseline to relapse-refractory states. Nat Commun. 2022;13:4517. DOI: 10.1038/s41467-022-32145-y.
23. Landau HJ, Yellapantula V, Diamond BT y col. Accelerated single cell seeding in relapsed multiple myeloma. Nat Commun. 2020;11:3617. DOI: 10.1038/s41467-020-17459-z.
24. Walker BA, Mavrommatis K, Wardell CP y col. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159-70. DOI: 10.1038/s41375-018-0196-8.
25. D’Agostino M, Cairns DA, Lahuerta JJ y col. Second Revision of the International Staging System (R2-ISS) for Overall Survival in Multiple Myeloma: A European Myeloma Network (EMN) Report Within the HARMONY Project. J Clin Oncol. 2022;40:3406-18. DOI: 10.1200/JCO.21.02614.
26. Harding T, Baughn L, Kumar S, Van Ness B. The future of myeloma precision medicine: integrating the compendium of known drug resistance mechanisms with emerging tumor profiling technologies. Leukemia. 2019;33:863-83. DOI: 10.1038/s41375-018-0362-z.
27. Dutta AK, Alberge JB, Sklavenitis-Pistofidis R, Lightbody ED, Getz G, Ghobrial IM. Single-cell profiling of tumour evolution in multiple myeloma — opportunities for precision medicine. Nat Rev Clin Oncol. 2022;19:223-36. DOI: 10.1038/s41571-021-00593-y.
28. Vo JN, Wu YM, Mishler J y col. The genetic heterogeneity and drug resistance mechanisms of relapsed refractory multiple myeloma. Nat Commun. 2022;13:3750. DOI: 10.1038/s41467-022-31430-0.
29. John L, Krauth MT, Podar K, Raab MS. Pathway-Directed Therapy in Multiple Myeloma. Cancers. 2021;13:1668. DOI: 10.3390/cancers13071668.
30. Giesen N, Chatterjee M, Scheid C y col. A phase 2 clinical trial of combined BRAF/MEK inhibition for BRAF V600E-mutated multiple myeloma. Blood. 2023;141:1685-90. DOI: 10.1182/blood.2022017789.
31. Kostopoulos IV, Ntanasis-Stathopoulos I, Gavriatopoulou M, Tsitsilonis OE, Terpos E. Minimal Residual Disease in Multiple Myeloma: Current Landscape and Future Applications With Immunotherapeutic Approaches. Front Oncol. 2020;10:860. DOI: 10.3389/fonc.2020.00860.
32. Zah E, Nam E, Bhuvan V y col. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat Commun. 2020;11:2283. DOI: 10.1038/s41467-020-16160-5.
33. Zhang X, Zhang H, Lan H, Wu J, Xiao Y. CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Front Immunol. 2023;14:1101495. doi: 10.3389/fimmu.2023.1101495. DOI: 10.3389/fimmu.2023.1101495.
34. Kropivsek K, Kachel P, Goetze S y col. Ex vivo drug response heterogeneity reveals personalized therapeutic strategies for patients with multiple myeloma. Nat Cancer. 2023;4:734-53. DOI: 10.1038/s43018-023-00544-9.
35. Boakye Serebour T, Cribbs AP y col. Overcoming barriers to single-cell RNA sequencing adoption in low-and middle-income countries. European Journal of Human Genetics. 2024;2:1-8. DOI: 10.1038/s41431-024-01564-4.