Linfoma de células T paniculítico: a propósito de un caso
Paniculitic T-cell lymphoma: about a case
Tarqui M, Vargas L, Gregorio C, Rosero JM, Sánchez J, Enrico A, Ponzinibbio C.
Hospital Italiano La Plata
ORCID:
Tarqui M 0009-0007-8945-6555
Vargas L 0009-0005-4551-400X
Gregorio C 0009-0002-6690-2827
Rosero JM 0009-0001-3416-2644
Sánchez J 0009-0000-1764-4951
Enrico A 0000-0002-1983-6145
Ponzinibbio C 0009-0000-8914-4219
Palabras claves: linfoma,
células T,
fiebre.
Keywords: lymphoma,
T cells,
fever.
Resumen
El linfoma de células T paniculítico representa menos del 1% de todos los linfomas no Hodgkin. Se distingue por placas y nódulos subcutáneos y es más frecuente en mujeres. El curso clínico puede ser desde indolente hasta agresivo asociado a síndrome hemofagocítico, presentando mal pronóstico y respuesta pobre al tratamiento. En este reporte de caso clínico hacemos mención a un paciente con linfoma de células T paniculítico cuyo único síntoma inicial fue fiebre prolongada que posteriormente agregó leucopenia y la aparición de nódulos de pequeño tamaño en miembros superiores.
Abstract
Panniculus T-cell lymphoma represents less than 1% of all non-Hodgkin's lymphomas, it is distinguished by subcutaneous plaques and nodules, most frequent in women, it can be indolent, or present an aggressive clinical course associated with hemophagocytic syndrome. It has a poor prognosis with poor response to treatment. In this report we will comment on the case of a patient with panniculitic T-cell lymphoma, whose only initial symptom was prolonged fever that later added leukopenia and the appearance of small nodules in the upper limbs.
El linfoma T cutáneo representa aproximadamente del 75 al 80% de todos los linfomas cutáneos primarios. El linfoma de células T tipo paniculítico (LSCT) es un linfoma primario postímico infrecuente, menos de 1% de todos los linfomas no Hodgkin. Fue descrito por primera vez por González y colaboradores en 1991, jerarquizando por sus características histopatológicas y un curso clínico agresivo (1). Se distingue clínicamente por placas y nódulos subcutáneos eritematosos que aparecen preferentemente en las extremidades inferiores y en el tronco. Es más frecuente en mujeres en la tercera y cuarta década de la vida, aunque también puede afectar a hombres y niños (2,3). Afecta preferentemente al tejido celular subcutáneo (TCS), y las células que lo constituyen expresan habitualmente el fenotipo de los receptores TCR-α/β o más raramente TCR-γ/δ(1,3). Este subtipo de linfoma tiene un curso indolente. Sin embargo si se asocia a síndrome hemofagocítico presenta mal pronóstico y baja respuesta al tratamiento (3). Exponemos el caso de un paciente de 19 años de edad, con diagnóstico de linfoma T paniculítico que presentó fiebre persistente asociado a leucopenia y pequeños nódulos subcutáneos en miembros superiores.
Caso clínico
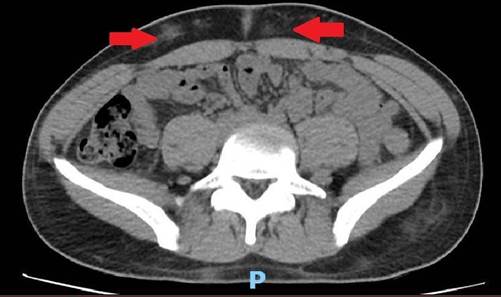
Paciente masculino de 19 años sin antecedentes personales patológicos, que fue derivado por síndrome febril prolongado de dos meses de evolución asociado a leucopenia, tratado previamente con antibioticoterapia empírica con resolución temporal de los registros febriles. Examen físico sin adenopatías ni organomegalias Estudio hematológico de sangre periférica: Hb 13.7 g/dL, Hto 43%, leucocitos 2.200/mm3 (60% pmn., 30% linf., 10% mo), plaquetas 288.000/mm3 Lab: ERS: 10 mm, PCR: 7 mg/dl (VN< 5), LDH: 1100 UI/L, ferritina: 679 mg/L (VN < 365). Se solicitó ecocardiograma normal. Se realizó tomografía con hallazgo de adenomegalias a nivel de ambos huecos axilares (16 mm), múltiples áreas hiperdensas a nivel del TCS en extensión toraco-abdominal anterior y posterior. (Figuras 1 y 2).
Se realizó PAMO + CMF - biopsia + mielocultivos. En el aspirado de médula ósea se observaron cambios megaloblastoides moderados en serie roja y ligera displasia granulocítica, con disminución de la diferenciación final.
La citometría de flujo informo que en la subpoblación T se detectó un desbalance en la relación CD4/CD8.
El paciente continuó con registros febriles, sin patrón específico y sin cambios en el estudio hematológico de sangre periférica (EHSP). Ferritina, LDH y PCR en ascenso. Mielocultivo: negativo. Se realizó interconsulta con Reumatología, que solicitó FAN, FR, C3-C4: negativos. Serología brucelosis: negativa
En el día +7 intercurrió con edemas en ambos brazos se solicitó ecografía con hallazgo de aumento de la ecogenicidad TCS de etiología inflamatoria/infecciosa, y celulitis (Figura 3), por lo que se decidió la realización de biopsia + cultivo de TCS. Posteriormente inició tratamiento empírico con corticoides por sospecha etiológica autoinmune. Se obtuvieron resultados de cultivo de TCS negativo. El paciente presentó mejoría de curva térmica e incremento en recuento leucocitario. Se otorgó alta hospitalaria con corticoides a la espera de resultado de biopsia TCS y ANCA.
Se recibió biopsia del TCS: reactividad de membrana para CD45 y CD3 en células linfoides KI67 60%. Presencia de macrófagos CD4+, linfocitos B (CD20+) escasos. Diagnóstico: infiltración intersticial linfoide hipodérmica compatible con LNH IHQ: LNH T cutáneo (CD3+, CD8+, granzime B+) tipo paniculitis (WHO 2016).
Ya con un diagnóstico filiado se recibió el reporte de la biopsia de médula ósea sin infiltración, levemente hipocelular con dishemopoyesis. El paciente presentó mejoría de la curva térmica con inicio de corticoides. Sin embargo las lesiones nodulares en miembros superiores presentaron mejoría parcial, por lo que se añadió ciclosporina al esquema terapéutico. Actualmente el paciente se encuentra en seguimiento con evolución favorable sin intercurrencias.
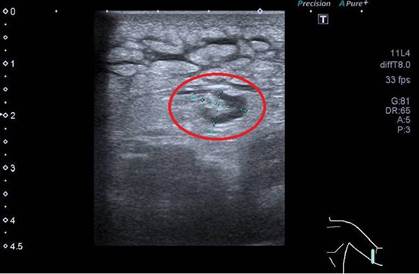 |
Figura 1.
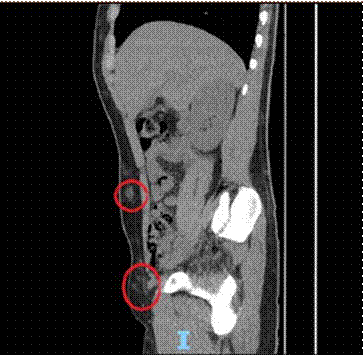 |
Figura 2.
Figura 3.
 |
Discusión
El LSCT es una variante distintiva de los linfomas cutáneos de células T, que se presenta como nódulos subcutáneos de apariencia inflamatoria, histológicamente constituidos por un infiltrado denso de linfocitos de tamaño mediano a grande, de apariencia atípica, hipercromáticos y con núcleos angulados con un patrón de proliferación predominantemente lobulillar, aunque pueden entrar en distribución septal y rodear a las células adiposas del TCS. Se pueden hallar entremezclados algunos histiocitos benignos, células plasmáticas o neutrófilos en los lóbulos (1,2). Se clasificaba en dos subtipos diferentes: un LSCT de curso indolente (82%), parecido a una paniculitis histiocítica citofágica, con fenotipo de células T TCR α/β citotóxicas, CD8+, CD56–, y supervivencia 5-10 años, y un LSCT de curso agresivo (18%), fenotipo TCR γ/δ citotóxico, CD8–, CD56+, EBV+, con supervivencia de un año (3).
Sin embargo, desde la reunión de la EORTC en 2004, se considera sólo LSCT aquellos casos con fenotipo habitualmente CD3+, CD4–, CD8+, CD30–, CD56–, y TCR α/β restringidos al tejido celular subcutáneo, en los que el síndrome hemofagocítico es poco frecuente y que, en general, presentan un curso indolente, con supervivencia a los 5 años del 80%. El segundo subtipo de LSCT, con TCR γ/δ se engloba ahora dentro de un grupo provisional aparte dentro de los linfomas T cutáneos (3).
Normalmente, el LSCT afecta a adultos jóvenes entre 30 y 50 años, aunque se han descrito casos en niños. Del total de los pacientes reportados, más de la mitad son mujeres.
Clínicamente se manifiesta por la presencia de nódulos o placas, únicos o múltiples, usualmente no ulcerados (80%). Se localiza principalmente en miembros inferiores y con menor frecuencia en tronco, extremidades superiores y cara (3). Las lesiones pueden regresar de forma espontánea o persistir. Se han descrito casos progresivos o con complicaciones sistémicas concomitantes como la hemofagocitosis (4). El edema facial como característica de presentación de LSCT ha sido también descrito en otros reportes de casos quienes sugieren que cualquier lesión cutánea atípica con aparente componente inflamatorio subcutáneo deben incluir como diagnóstico diferencial el LSCT (5).
En raras ocasiones, LSCT también imita la dermatomiositis, mostrando asimismo una erupción de heliotropo como debilidad muscular proximal (6). Nuestro caso presentó lesiones nodulares eritematosas en brazos.
El diagnóstico diferencial de esta entidad es en ocasiones difícil por la localización atípica del infiltrado linfoide, además de la clínica inespecífica y la presencia de síntomas sistémicos. Se deben excluir siempre las paniculitis reactivas o las formas autoinmunitarias, como la paniculitis lúpica y la dermatomiositis. En cuanto a la paniculitis lúpica, su presentación clínica es generalmente difícil de diferenciar. Algunas claves que ayudan en su diferenciación son la presencia de células plasmáticas en la paniculitis lúpica y los agregados nodulares de células B que forman centros germinales en la periferia de los lóbulos, mientras que, en los estudios moleculares, en el linfoma de células T tipo paniculítico se evidencia, la mayoría de las veces, la presencia de un reordenamiento clonal (7). Por otro lado, muchos pacientes que han sido diagnosticados de paniculitis histiocítica citofágica son, en realidad, verdaderos LSCT, ya que estas patologías se solapan (1-3).
El tratamiento en esta patología es controvertido, basado en el fenotipo y en la agresividad de la enfermedad. La mayoría de veces es suficiente el manejo con esteroides sistémicos a 1 mg/kg/día. Puede combinarse con hidroxicloroquina o colchicina con adecuada reacción terapéutica en algunos casos si asociado a una observación estricta (3,7). Se han reportado también casos de reacción terapéutica completa y largas tasas de remisión, con ciclosporina como monoterapia o en combinación con metotrexato (7,8). En las formas agresivas, resistentes o asociadas al síndrome hemofagocítico, se hace necesario el manejo con poliquimioterapia asociada a radioterapia, con esquemas tipo CHOP o variaciones. En casos resistentes al tratamiento, el trasplante alogénico de médula ósea podría llegar a ser de utilidad, aunque se ha reportado una tasa de supervivencia de tan sólo dos años y debería reservarse únicamente para la enfermedad progresiva con afectación extracutánea (7). En nuestro paciente se inició tratamiento con corticoides que mejoraron la curva térmica, pero ante la persistencia de lesiones cutáneas y confirmación de diagnóstico se decidió iniciar tratamiento con ciclosporina presentando buena respuesta. El pronóstico de la entidad es variable; sin embargo, se considera una entidad lentamente progresiva, con buen pronóstico y supervivencia a cinco años de más del 50%. Sin embargo, en presencia de síndrome hemofagocítico se asocia a mala respuesta al tratamiento con pronóstico pobre (3,7).
Conclusiones
El LSCT es un tipo de linfoma raro, en ocasiones agresivo. El diagnóstico suele ser complicado por los síntomas inespecíficos y sistémicos que pueden ser solapados con otras entidades, pudiendo debutar como una paniculitis inespecífica o, como en nuestro caso, con un síntoma inespecífico como la fiebre. Dada la baja frecuencia de presentación y la escasa bibliografía, muchos autores recomiendan que se tenga en cuenta la entidad ante la presencia de lesiones atípicas, continuando, de ser conveniente, con biopsias secuenciales y estudios inmunohistoquímico, genéticos y moleculares completos.
Bibliografía
1. Marzano AV, Berti E, Pauli M et al. Cytophagic histiocytic panniculitis and subcutaneous panniculitis-like T-cell lymphoma. Report of 7 cases. Arch Dermatol. 2000; 136:889-896.
2. Wick MR, Patterson JW. Cytophagic histiocytic panniculitis a critical reappraisal. Arch Dermatol. 2000;136:922-924.
3. Rodriguez-Vasquez M, Garcia-Arpa M, Martin F et al. Linfoma de células T paniculítico. Actas Dermo-Sifiliográficas, 2005;96(2):98-101.
4. Smith KJ, Skelton HG, Yeager J y col. Cutaneous histopathologic, immunohistochemical, and clinical manifestations in patients with hemophagocytic syndrome. Arch Dermatol. 1992;128:193-200.
5. Yung A, Snow J, Jarrett P. Subcutaneous panniculitic T-cell lymphoma and cytophagic histiocytic panniculitis. Aust J Derm. 2001;42:183-187.
6. Prescott RJ, Banerjee SS, Cross PA. Subcutaneous T-cell lymphoma with florid granulomatous panniculitis. Histopathology. 1992;20:535-537.
7. Giraldo A, Yurledy Del Rio D., Ruiz, A. Linfoma cutáneo paniculítico de células T asociado a síndrome hemofagocítico, a propósito de un caso. Rev Asoc Colombia Dermatol. 2015;24(1):47-50.
8. Lee WS, Hwang J-H, Kim MJ, Go S-I, Lee A, Song H-N et al. Cyclosporine A as a primary treatment for panniculitis-like T cell lymphoma: A case with a long-term remission. Cancer Res Treat. 2014;46:312-6.