Macroglobulinemia de Waldenström. Estudios citogenéticos y moleculares
Waldenström macroglobulinemia. Cytogenetic and molecular studies
Stanganelli C1, Cabrera J1, Slavutsky I2
1 División Patología Molecular, Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina, Buenos Aires, Argentina.
2 Laboratorio de Genética de Neoplasias Linfoides, Instituto de Medicina Experimental, CONICET-Academia Nacional de Medicina, Buenos Aires, Argentina
Palabras clave: macroglobulinemia de Waldesnström,
MYD88,
CXCR4,
mutaciones.
Key words: Waldenström's macroglobulinemia,
MYD88,
CXCR4,
mutations.
Resumen
La macroglobulinemia de Waldenström (MW) es un linfoma linfoplasmocítico con compromiso de la médula ósea (MO) y presencia de una gammapatía monoclonal IgM. La alteración citogenética más frecuente es la deleción de parte del brazo largo del cromosoma 6 observada en el 30-54% de los casos, asociada a factores de pronóstico adverso en esta patología. Los estudios de secuenciación masiva permitieron detectar la presencia de mutaciones de los genes MYD88 y CXCR4, de valor diagnóstico y pronóstico en esta entidad. La mutación activante del gen MYD88, que determina el cambio del aminoácido leucina por prolina en la posición 265 de la proteína (MYD88L265P), se observa en el 93-97% de los pacientes con MW y en el 40-60% de los casos de gammapatía monoclonal de significado incierto (MGUS) IgM, en tanto que las mutaciones de CXCR4 se encuentran en el 30-40% de los pacientes con MW, siendo menos frecuentes en el MGUS-IgM (4-20%). CXCR4S338X es la variante más común (50% de las mutaciones); genera un codón stop que conduce a una proteína truncada en el aminoácido 338, y a la pérdida de 15 aminoácidos en el dominio C-regulador. Algunos pacientes presentan mutaciones múltiples en distintos subclones. Los casos con MYD88WT/CXCR4WHIM/WT tienen pronóstico adverso con corta sobrevida libre de progresión y global. Los pacientes con MYD88L265P/CXCR4WHIM/WT tienen buena respuesta al tratamiento, en tanto que aquéllos con ambos genes mutados presentan un pronóstico intermedio. Sin duda, el análisis de estas mutaciones ha permitido profundizar la caracterización biológica de la MW haciendo factible en un futuro ampliar la posibilidad de disponer de nuevos blancos terapéuticos.
Abstract
Waldenström macroglobulinemia (WM) is a lymphoplasmacytic lymphoma with bone marrow (BM) involvement and the presence of a monoclonal IgM gammopathy. The most frequent cytogenetic alteration is the deletion of part of the long arm of chromosome 6 observed in 30-54% of cases, associated with adverse prognostic factors in WM. The introduction of next generation sequencing allowed the detection of mutations in the MYD88 and CXCR4 genes, with diagnostic and prognostic value in this pathology. The activating mutation of the MYD88 gene determines the change of the amino acid leucine for proline at position 265 of the protein (MYD88L265P) that was observed in 93-97% of patients with WM and in 40-60% of cases of monoclonal gammopathy of undetermined significance (MGUS) IgM. CXCR4 mutations were found in 30-40% of patients with WM, being less frequent in MGUS-IgM (4-20%). The most common variant, CXCR4S338X (50% of mutations), generates a stop codon leading to a truncated protein at amino acid 338 and to the loss of 15 amino acids in the C-regulatory domain. Some patients have multiple mutations in different subclones. Cases with MYD88WT/CXCR4WHIM/WT have the worst prognosis with short overall and progression-free survival. Patients with MYD88L265P/CXCR4WHIM//WT have a good response to treatment, whereas those with both mutated genes have an intermediate prognosis. Undoubtedly, the analysis of these mutations has allowed the increase of the biological characterization of WM, making possible in the future to expand the probability of having new therapeutic targets.
Introducción
La Organización Mundial de la Salud define a la macroglobulinemia de Waldenström (MW) como un linfoma linfoplasmocítico (LPL) con compromiso de la médula ósea (MO) y la presencia de una gammapatía monoclonal IgM de cualquier concentración(1,2). La MW corresponde al 95% de los LPL (<5% son secretores de IgG, IgA o no secretores) y presenta un patrón de infiltración de la MO predominantemente intratrabecular con un incremento en el número de mastocitos. Es una enfermedad rara, que representa aproximadamente el 1-2% de las neoplasias hematológicas, con una incidencia aproximada de 0,57/100000 personas por año(3,4), siendo mayor en caucásicos (0,41/100000 por año) que en afrodescendientes (0,18/100000 por año)(5). La edad media de presentación es 63-68 años con predominio del sexo masculino. Los pacientes menores de 70 años tienen una media de sobrevida (SV) mayor de 10 años, aquéllos entre 70 y 79 años de aproximadamente 7 años y los mayores de 80 años de alrededor de 4 años(6). La mayoría de los casos se originarían a partir de una célula B de memoria caracterizada por un fenotipo CD27+, IgM+, IgD-, CD25+ CD22+low, arrestada después de sufrir hipermutación somática en el centro germinal y antes de alcanzar la diferenciación a célula plasmática(7-10).
El principal factor de riesgo de desarrollo de una MW es la preexistencia de MGUS (monoclonal gammopathy of undetermined signficance) IgM, seguido de la presencia de una historia familiar de MW u otra neoplasia linfoide a células B y factores inmunológicos. En particular, el MGUS-IgM confiere un riesgo relativo 46 veces más alto que el de la población general de desarrollar MW(11). Si bien en la mayoría de los casos es una enfermedad esporádica, diferentes trabajos refieren agregación familiar, con un riesgo hasta 20 veces superior en los familiares de primer grado de presentar MW u otro desorden linfoproliferativo respecto de la población general(12,13), sugiriendo una susceptibilidad genética en el desarrollo de esta entidad. Asimismo, diferentes reportes detectan menor edad de comienzo de la enfermedad, mayor nivel de infiltración de la MO y menos sobrevida en los casos con historia familiar de MW o de algún proceso linfoproliferativo(14,15).
Los síntomas clínicos más comunes son debilidad y fatiga, generalmente secundarios a anemia, y síntomas B (pérdida de peso, sudoración excesiva y fiebre baja), y afectan a una cuarta parte de los pacientes. En el 15-30% de los casos se observa hepatomegalia, esplenomegalia y linfadenopatía(4,16-18). Asimismo, los niveles elevados de IgM circulante producen manifestaciones clínicas vinculadas a las propiedades fisicoquímicas de la proteína monoclonal IgM, como hiperviscosidad, neuropatía periférica, crioglobulinemia o enfermedad de aglutininas frías(4,19). El depósito de agregados amorfos de IgM se asocia a disfunción orgánica, particularmente a nivel gastrointestinal y renal, así como a alteraciones en la piel. En cuanto al inmunofenotipo, las células de la MW expresan CD19, CD20, CD22low, CD25, CD27, CD79b, CD81, FMC7, IgMs, son CD5, CD10, CD11c y CD103 negativas(1,10), y presentan restricción de cadena liviana κ o λ con una relación 5:1(20), siendo de importancia para distinguir una MW de otras neoplasias linfoides.
En referencia al gen IGHV (immunoglobulin heavy chain variable region), el mismo se encuentra fuertemente mutado en la mayoría de los casos de MW y MGUS-IgM, con un rango de desviación de la línea germinal de 2,1% a 16,2%. La comparación con el repertorio de genes presentes en células B normales, evidencia una sobre-representación de la familia VH3, baja frecuencia de VH1 y VH4, asociado a un uso sesgado de genes IGHV, con aumento de expresión de IGHV3-23, IGHV3-64, IGHV3-7 y IGHV3-74, y disminución de IGHV4-39. Estos datos, en concordancia con el inmunofenotipo, sustentan la hipótesis de que los eventos transformantes ocurren por presión selectiva en células de memoria post centro germinal(21-23). En cuanto al análisis de la secuencia de aminoácidos en la región VH CDR3, a diferencia de lo encontrado en leucemia linfocítica crónica (LLC), la MW no evidencia la presencia de subsets o grupos de homología que permitan identificar receptores estereotipados(22, 24).
Alteraciones citogenéticas
Al presente, y en comparación con otros desórdenes linfoproliferativos, es poca la información existente respecto de las anomalías cromosómicas observadas en la MW debido, fundamentalmente, al bajo índice mitótico de las células tumorales in vitro. No obstante, si bien esta patología no presenta alteraciones específicas, la frecuencia de las anomalías encontradas difiere de lo observado en otras neoplasias linfoides. Asimismo, el análisis de CNA (copy number aberrations) muestra una media de 4 alteraciones por caso, similar a lo observado en LLC o linfomas de la zona marginal, pero mucho menor que lo detectado en el linfoma de células del manto o en el mieloma múltiple(25,26).
En particular, la alteración más frecuente es la deleción (del) de parte del brazo largo del cromosoma 6 (del6q) observada en el 30-54% de los casos, seguida por: trisomía 18 (15-23%), del13q (13-15%), del17p (8-23%), trisomía 4 (8-12%), del11q (7%) y trisomía 12 (<5%)(26-31). Nguyen-Khac y col(31) observaron una asociación significativa entre las trisomías 4 y 18. Simultáneamente, son muy poco frecuentes las translocaciones que involucran el gen de la cadena pesada de las inmunoglobulinas (IGH) (3%)(31,32). En cuanto a su significado clínico, las deleciones 6q y 11q, así como la trisomía 4 se asocian a factores de pronóstico adverso en la patología, pero no presentan impacto sobre su evolución clínica(31,33,34), en tanto que la del17p y la trisomía 12 se relacionan a una corta sobrevida libre de progresión (SLP)(31).
En referencia a la del6q, se han descripto dos regiones de mínima deleción: 6q21 y 6q23, que determinan la pérdida de genes con importantes funciones regulatorias que modulan la actividad de NF-kB (TNFAIP3, HIVEP2), apoptosis (FOXO3), proteínas de la familia BCL2 (BCLAF1), BTK (IBTK) y diferenciación plasmocítica (PRDM1, ARID1B)(26,35-37). Un trabajo reciente(38) compara los perfiles de expresión de pacientes con y sin del6q, observando que los casos con la deleción presentan aumento de la expresión de los genes de la vía de señalización del receptor de células B (BCR; B-cell receptor) (CD79a, SYK, BLNK, PLC𝛾2, CARD11) y de IL-2 (interleukin 2). Los autores postulan que la activación del BCR estaría posiblemente asociada a la pérdida de BLIMP-1 (B- lymphocyte-induced maturation protein 1) (6q21) cuya función en condiciones normales se encuentra relacionada con la inhibición de la proliferación y activación de los linfocitos B, incluyendo el camino de señalización del BCR, así como con la diferenciación de las células plasmáticas(39). Por su parte, en MW, IL-2 contribuye a la secreción de IgM y a la proliferación celular vía la activación del camino de señalización JAK/STAT (Janus kinasa/Signal transducer and activator of transcription proteins)(40).
Rearreglos moleculares
Mutaciones de MYD88 (myeloid differentiation primary response 88)
Estudios de secuenciación masiva de última generación (NGS; next generation sequencing) permitieron detectar una mutación activante en el gen MYD88, ubicado a nivel de 3p22.2, que determina el cambio del aminoácido leucina por prolina en la posición 265 de la proteína (L265P) (MYD88L265P)(41). La misma se observa en aproximadamente el 93-97% de los pacientes con MW y en el 40-60% de los casos de MGUS-IgM, dependiendo de la metodología empleada para su detección(35,41). El gen MYD88 codifica una proteína adaptadora citosólica que desempeña un papel central en la respuesta inmune innata y adaptativa. Dicha proteína funciona como un transductor de señal esencial en las vías de señalización de los receptores de interleuquina-1 y de tipo Toll(41). MYD88L265P induce la activación de las kinasas IRAK (interleukin-1 receptor–associated kinase) y BTK (Bruton’s tyrosine kinase), llevando a la activación de NF-kB (nuclear factor- kB) y al desarrollo neoplásico(42,43) (Figura 1). El reclutamiento y activación de las moléculas IRAK y BTK puede ser bloqueado por inhibición de MYD88, llevando a la apoptosis de las células neoplásicas con MYD88 mutado. La ausencia de mutación (MYD88WT) se asocia a mayor edad, menor infiltración de la MO y compromiso extramedular, un curso clínico más agresivo con menor SV, así como un riesgo de muerte diez veces más alto que los casos portadores de MYD88L265P (36,44,45). Asimismo, los pacientes con MYD88WT pueden presentar mutaciones somáticas recurrentes que impactan en la vía de señalización de NF-kB, en reguladores epigenómicos y en los genes DDR (DNA damage responsive) así como una alta incidencia de linfoma difuso de grandes células B (LDGCB)(37,46). Las mutaciones más comunes afectan la vía de señalización de NF-kB e incluyen los genes TBL1XR1, NFKBIB, NFKBIZ, NFKB2, MALT1, BCL10 y UDRLIF, y el complejo de genes CBM (CARD11-BCL10-MALT1). Adicionalmente, se han identificado mutaciones diferentes de la L265P, que incluyen S219C, M232T y S243N, aunque sus frecuencias son mucho más bajas (1-2%)(45).

Figura 1. Vías metabólicas de MYD88 y de CXCR4 en macroglobulinemia de Waldenström. Se detallan las mutaciones somáticas encontradas con mayor frecuencia en el extremo c-terminal de CXCR4. fs: frameshift. Modificada de Castillo et al(43).
En cuanto a la metodología, en los pacientes con sospecha de MW, el análisis por ASO-PCR (PCR-alelo específica) en MO representa el ensayo de elección para la detección de la mutación MYD88L265P (22,47,48). No obstante, la misma puede también detectarse por ASO-PCR en sangre periférica de pacientes sin tratamiento previo(49). Como la sensibilidad disminuye en los pacientes tratados, en estos casos se recomienda usar sólo muestras de MO para identificar de manera confiable la mutación(13,36). En pacientes con MGUS-IgM, Varettoni y col.(36) observaron resultados discordantes en la identificación de la mutación MYD88L265P entre las técnicas de NGS y PCR en tiempo real alelo específica (Taqman Allele-Specific Genotyping Assay), detectándose la mutación sólo con la última metodología, que tendría mayor sensibilidad diagnóstica cuando el clon de células B es pequeño(36).
Simultáneamente, en MW no se observaron diferencias en el porcentaje de casos mutados efectuando el análisis en células mononucleares totales de MO respecto de células CD19+ seleccionadas(36,50). Otra alternativa para la detección confiable de la mutación MYD88L265P y el monitoreo de enfermedad mínima residual es el método de ddPCR (droplet digital PCR), de sensibilidad superior a la ASO-PCR, especialmente útil para el estudio de muestras con bajo porcentaje de infiltración, tales como MO no seleccionadas o sangre periférica. La PCR digital podría utilizarse también para la detección de la mutación MYD88L265P en ADN tumoral circulante en plasma(51).
Resulta interesante destacar que las alteraciones estructurales del brazo corto del cromosoma 3 pueden modificar la carga alélica de MYD88L265P, debido a la deleción del alelo WT (WT: wild type o no mutado), amplificación del alelo mutado o disomía uniparental adquirida resultando en homocigocidad(35,41). Esta mutación también se ha observado, aunque con frecuencias más bajas, en otras neoplasias hematológicas, como LDGCB, linfoma de tejido linfoide asociado a la mucosa (MALT) y LLC(41,47,52,53), pero está ausente en el mieloma múltiple(41).
Asimismo, se ha encontrado asociación entre la presencia de MYD88L265P y la del6q, sugiriendo roles compartidos de ambos eventos genómicos. En este aspecto, los casos con las dos alteraciones muestran con mayor frecuencia pérdida del gen BCLAF1 (BCL2 Associated Transcription Factor 1), seguido por TNFAIP3, HIVEP2, IBTK, y FOXO3. Simultáneamente, el análisis de la región comprendida entre las bandas 6q14 y 6q27 muestra dos patrones diferentes de deleción: el 40% de los casos presenta pérdidas clonales y contiguas abarcando todos los genes involucrados en la región, en tanto que el 60% restante tiene deleciones más focales y subclonales(37).
Por otra parte, sabemos que el riesgo de progresión de MGUS-IgM a MW u otros desórdenes linfoproliferativos es de 1,5% por año(54) y que tanto la presencia de la mutación MYD88L265P como su nivel de expresión están asociadas a la transformación maligna(53,55). Considerando que la progresión de MGUS-IgM (condición premaligna) a MW (neoplasia) ocurre en un proceso de múltiples pasos, la alta prevalencia de MYD88L265P en MGUS-IgM confirma que esta mutación es un evento oncogénico temprano. Por su parte, la presencia de otras mutaciones y/o CNA determinaría una expresión génica anormal que promovería la progresión de la enfermedad(35).
Mutaciones de CXCR4 (C-X-C Motif Chemokine Receptor 4)
Además de la mutación del gen MYD88, estudios de NGS mostraron la presencia de mutaciones en el gen CXCR4, localizado en 2q22, en el 30-40% de los pacientes con MW, siendo menos frecuentes en MGUS-IgM (4-20%)(35,36,42,56,57). Las mutaciones de CXCR4 son esencialmente exclusivas de la MW, ya que no se han descrito hasta ahora en otras enfermedades, con la excepción de unos pocos linfomas de células B de zona marginal y casos de LDGCB subtipo ABC (Activated B-cells). La ubicación de las mutaciones somáticas en el dominio C terminal en pacientes con MW es similar a lo observado en la línea germinal de pacientes con síndrome de WHIM (Warts, Hypogammaglobulinaemia, Infections and Myelokathexis), una inmunodeficiencia congénita caracterizada por neutropenia crónica no cíclica(58).
CXCR4 es un receptor de quemoquinas que promueve la sobrevida, migración y adhesión de las células tumorales al estroma de la MO a través de interacciones con el ligando CXCL12(59). En condiciones normales, después de la unión a su ligando, CXCR4 se activa, se une a proteínas G y se producen una serie de eventos que activan la tirosina quinasa de la familia Src, seguido de PI3K (phosphatidylinositol-3-kinase), la vía JAK/STAT independientemente de las proteínas G, seguido de ERK (extracellular signal-regulated kinases) por β-arrestinas, causando la migración, adhesión y transcripción de genes. Después de su unión a CXCL12, CXCR4 se internaliza rápidamente, es ubiquitinado y degradado(60). Se han descripto más de 40 tipos diferentes de mutaciones somáticas en CXCR4 en pacientes con MW, incluyendo las variaciones con corrimiento del marco de lectura (CXCR4WHIM/FS) que comprometen una región de más de 40 aminoácidos del dominio C-terminal y mutaciones sin sentido (CXCR4WHIM/NS) que truncan 15-20 aminoácidos de la región distal(35,44) (Figura 1). Al igual que en el síndrome de WHIM, dichas mutaciones dejan intactas las siete hélices de transmembrana involucradas en la señalización y unión del ligando, pero resultan en el truncamiento de la cola citosólica de la proteína que contiene las fosfoserinas regulatorias, alterando la internalización y produciendo una activación prolongada(61). La variante más común es CXCR4S338X, que representa más del 50% de las mutaciones de este gen en pacientes con MW. Consiste en una transversión C>A o C>G en el nucleótido 1013 de CXCR4, que da como resultado la generación de un codón stop que conduce a una proteína truncada en el aminoácido 338, y la pérdida de 15 aminoácidos en el dominio C-regulador. Asimismo, entre las mutaciones CXCR4S338X, C>G es más frecuente que C>A(42). Ambas variantes sin sentido (CXCR4S338X C>A y C>G) se asocian con formas más agresivas de la enfermedad al diagnóstico. Los pacientes con MYD88L265P/CXCR4WHIM/NS tienen mayor compromiso de la MO y altos niveles séricos de IgM, más riesgo de hiperviscosidad, mayor requerimiento de tratamiento, y son más propensos a padecer enfermedad de von Willebrand adquirido. Los casos con MYD88L265P/CXCR4WHIM/FS o MYD88L265P/CXCR4WHIM/WT presentan compromiso de la MO y niveles séricos de IgM intermedios, mientras que aquéllos con MYD88WT/CXCR4WHIM/WT (5-10% de los pacientes) tienen el peor pronóstico con baja infiltración de la MO y corta SLP y global. Los pacientes con MYD88L265P/CXCR4WHIM/NS o FS tienen pocas adenopatías y menores niveles séricos de β2 microglobulina respecto de aquéllos con MYD88L265P/CXCR4WHIM/WT (44,62-65). Las mutaciones del gen CXCR4 acortan la sobrevida libre de tratamiento (SLT) de los pacientes con MW asintomática, pero no tienen efecto en la SV global. Por su parte, la mutación MYD88L265P no influencia la SLT(44,65).
Las técnicas de secuenciación de Sanger o NGS son la mejor opción para el análisis del gen CXCR4 debido a la variedad de mutaciones reportadas en el mismo; sin embargo, en pacientes con baja carga tumoral en MO pueden obtenerse falsos-negativos. Dado que la mutación CXCR4S338X es la más frecuente, su búsqueda con la técnica de ASO-PCR mejora la sensibilidad diagnóstica para esta mutación en particular(56), pero debe combinarse con la secuenciación de Sanger que permite detectar las otras mutaciones posibles de encontrar en un mismo paciente(66). Hasta el presente, el método de detección de mutaciones de CXCR4 no ha sido estandarizado, sin embargo, en todos los casos la condición ideal incluye pacientes no tratados, uso de células mononucleares de MO y selección de células CD19+(42). Asimismo, la mutación CXCR4S338X se ha detectado con alta eficiencia por ASO-PCR en tiempo real en DNA tumoral circulante(67). Hunter y col(68) han demostrado que CXCR4WHIM disminuye la expresión de genes que se transcriben en respuesta a la mutación de MYD88.
El estudio de las mutaciones del gen CXCR4 por clonado y secuenciación en pacientes con MW reveló que la mutación es subclonal, en tanto que el análisis de la frecuencia alélica mostró que en la mayoría de los casos estarían presentes en el clon dominante. Asimismo, se observó la existencia de pacientes con múltiples mutaciones en subclones distintos y se detectó también la presencia de doble heterocigotas. Se observaron además clones homocigotas para una mutación determinada. La presencia de mutaciones subclonales en MW y su baja frecuencia en el MGUS-IgM revelan que las mismas ocurrirían después de la mutación MYD88L265P, durante la oncogénesis de la patología, aunque por su presencia en MGUS seguiría siendo un evento temprano(36,55). Simultáneamente, la detección de múltiples mutaciones de CXCR4 en un mismo paciente sería indicativa de inestabilidad genómica(36,42,66). Es importante destacar que, aunque usualmente los pacientes que tienen mutaciones del gen CXCR4 también portan la mutación MYD88L265P, pueden observarse pocos casos con MYD88WT y CXCR4 mutado (5%)(69).
Otras mutaciones
Además de las ya descriptas, se han detectado otras mutaciones en la MW, entre ellas las mutaciones somáticas en ARID1A (AT-rich interactive domain 1A) están presentes en el 17% de los casos e incluyen mutaciones puntuales que producen una proteína truncada o cambios con corrimiento del marco de lectura. Este gen puede modular TP53 y actuaría como un gen supresor epigenético(70). Asimismo, se observaron mutaciones de CD79A y CD79B en el 8% a 12% de los pacientes con MW, preferentemente en casos con MYD88MUT, aunque mutaciones en CD79B se encontraron también en pacientes con MYD88WT. CD79A y CD79B son componentes de la vía del BCR, ambos forman un heterodímero que se asocia con IGHV, proceso necesario para la expresión de superficie del BCR(71). Además, la pérdida del número de copias de LYN (LYN proto-oncogene) está presente en el 60% de los casos y puede favorecer la respuesta del BCR. Si bien las células B clonales de la MW exhiben figuras funcionales de activación crónica del BCR(72), la contribución de las mutaciones de CD79A y CD79B y las deleciones de LYN a la presentación clínica y respuesta al tratamiento debe ser evaluada en detalle.
Nuestra experiencia
En cuanto a nuestra experiencia, al presente efectuamos el análisis de mutaciones de los genes MYD88 y CXCR4 en una cohorte de 31 pacientes con MW: 22 al momento del diagnóstico, 4 en recaída y 5 durante el control post-tratamiento (13 varones; edad media 67,5 años, rango: 52-85 años) y 12 con MGUS-IgM (5 varones; edad media 76,9 años, rango: 68-88 años). En el 60% de los casos se efectuó la evaluación en muestras de MO, en tanto que en los restantes se empleó sangre periférica. Para el análisis de la mutación MYD88L265P se usó la técnica de ASO-PCR acorde a lo previamente descripto(47). Se realizaron dos reacciones de PCR que comparten el mismo primer sentido MYD88s, en una se amplificó la banda control de 160 pb que contiene el sitio L265P de la mutación empleando el primer antisentido MYD88as y en la otra se amplificó el alelo mutado de 122pb con el primer antisentido MYD88as-mut (Figura 2).
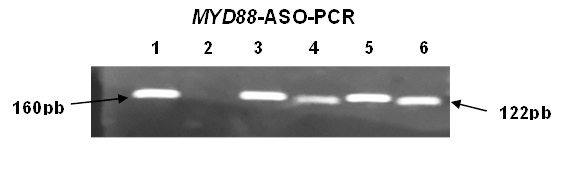
Figura 2. ASO-PCR de MYD88. Gel de agarosa al 2% mostrando una banda control de 160 pb (calles 1, 3 y 5) y una banda de 122 pb que amplifica al alelo mutado (calles 4 y 6). Calle 2: control negativo; calles 3 y 4: paciente con la mutación MYD88L265P; calles 5 y 6: control positivo para la mutación MYD88L265P.
La búsqueda de la mutación CXCR4S338X se efectúo en 22 pacientes con MW y 11 con MGUS-IgM mediante ASO-PCR según lo previamente reportado(42). Se utilizaron tres reacciones de PCR que comparten el mismo primer sentido CXCR4s. En la primera se amplificó el alelo normal con el primer antisentido CXCR4as, en la segunda se amplificó la mutación CXCR4S338X C>G con el primer antisentido CXCR4as C>G, y en la tercera se amplificó el alelo CXCR4S338X C>A con el primer antisentido CXCR4as C>A. En los tres casos se amplificaron fragmentos de 162 pb (Figura 3a). Para la detección de otras mutaciones del gen se utilizó la secuenciación bidireccional de Sanger(35). Los pacientes con MGUS evaluados resultaron negativos, 1 paciente con MW presentó la mutación CXCR4S338X C>G, y en un caso se detectó la mutación R334X, c.1000C<T (Figura 3b).
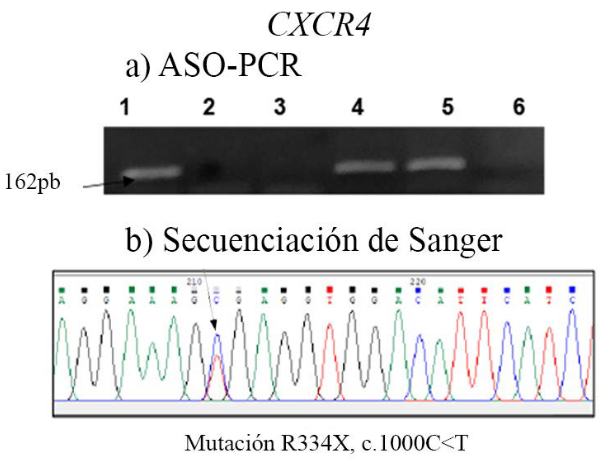
Figura 3. Análisis mutacional de CXCR4. a) ASO-PCR para la mutación CXCR4S338X. Gel de agarosa al 2%. Calles 1, 2 y 3: paciente negativo para las mutaciones C>G y C>A, se observa sólo la banda WT (wild type) (calle 1); calles 4, 5 y 6: paciente portador de la mutación C>G (calle 4: alelo WT, calle 5: alelo C>G positivo; calle 6: alelo C>A negativo). b) Secuenciación de Sanger mostrando la mutación R334X, c.1000C<T del gen CXCR4.
En la Tabla 1 se detallan los resultados obtenidos en los pacientes con MW en distintos estadios de la enfermedad y MGUS-IgM. El análisis de los datos mostró la siguiente distribución: MYD88MUT/CXCR4WT (85,7% de los casos), MYD88MUT/CXCR4MUT (9,5%) y MYD88WT/CXCR4WT (4,8%). No se encontraron mutaciones de CXCR4 en los pacientes con MGUS-IgM. Nuestra cohorte mostró positividad para MYD88 dentro de los valores reportados, en cambio encontramos una baja frecuencia de mutaciones en CXCR4. A nuestro entender, éste es el primer análisis de ambas mutaciones en pacientes con WM e MGUS-IgM de nuestro país.
Tabla 1. Distribución de mutaciones de MYD88L265P y CXCR4.
|
Pacientes |
MYD88L265P (%) |
CXCR4 (%) |
|
MW |
2/22 (9) |
|
|
- Al diagnóstico |
81,8 |
1/17 (5,9) |
|
- En recaída |
100 |
1/4 (25) |
|
- Control postratamiento |
0 |
0/1 (0) |
|
MGUS-IgM |
41,6 |
0/11 (0) |
Al presente existen distintos esquemas de tratamiento, siendo de destacar la utilización de nuevos agentes tales como inhibidores de BTK, PI3K, BCL2 y del proteasoma(56,63,73). Diferentes estudios muestran que el ibrutinib (inhibidor de BTK) resulta eficaz en el tratamiento de MW sintomática en pacientes MYD88L265P/CXCR4WHIM/WT. No obstante, se observa resistencia en los casos con mutaciones de CXCR4, particularmente en aquéllos con CXCR4WHIM/NS (46,74-77), mientras que los pacientes con CXCR4WHIM/FS no presentan diferencias respecto de los casos con CXCR4WHIM/WT (77). Estos datos muestran la importancia del análisis de la presencia de mutaciones en MW, tendiente a refinar el diagnóstico y pronóstico de esta patología, y profundizar la caracterización biológica de la enfermedad. La búsqueda de nuevas mutaciones permitirá ampliar en un futuro la posibilidad de disponer de nuevos blancos terapéuticos.
Agradecimientos
El presente trabajo se efectuó con del Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) y de la Fundación “Alberto J. Roemmers”, Buenos Aires, Argentina.
Bibliografía
1. Owen RG, Treon SP, Al-Katib A y col. Clinicopathological definition of Waldenström’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenström’s Macroglobulinemia. Semin Oncol. 2003; 30: 110-5.
2. Swerdlow SH, Campo E, Harris NL y col. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
3. Kyle RA, Larson DR, McPhail ED y col. Fifty-year incidence of Waldenström Macroglobulinemia in Olmsted County, Minnesota, from 1961 through 2010: a population-based study with complete case capture and Hematopathologic review. Mayo Clin Proc. 2018; 93: 739-46.
4. Gertz, MA. Waldenström macroglobulinemia: 2019 update on diagnosis, risk stratification, and management. Am J Hematol. 2019; 94: 266-76.
5. Wang H, Chen Y, Li F y col. Temporal and geographic variations of Waldenström macroglobulinemia incidence: a large population-based study. Cancer. 2012; 118: 3793-800.
6. Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenström macroglobulinaemia: an analysis of the surveillance, epidemiology and end results database. Br J Haematol. 2015; 169:81-9.
7. Sahota SS, Forconi F, Ottensmeier CH y col. Typical Waldenström macroglobulinemia is derived from a B-cell arrested after cessation of somatic mutation but prior to isotype switch events. Blood. 2002; 100:1505-7.
8. Kriangkum J, Taylor BJ, Strachan E y col. Impaired class switch recombination (CSR) in Waldenström macroglobulinemia (WM) despite apparently normal CSR machinery. Blood. 2006; 107: 2920-7.
9. Paiva B, Corchete LA, Vidriales MB y col. The cellular origin and malignant transformation of Waldenström macroglobulinemia. Blood. 2015; 125: 2370- 80.
10. García-Sanz R, Jiménez C, Puig N y col. Origin of Waldenström's Macroglobulinaemia. Best Pract Res Clin Haematol. 2016; 29: 136-47.
11. Ghobrial IM. Are you sure this is Waldenström macroglobulinemia? Hematology Am Soc Hematol Educ Program. 2012; 2012: 586-94.
12. Kristinsson SY, Bjorkholm M, Goldin LR y col. Risk of lymphoproliferative disorders among first-degree relatives of lymphoplasmacytic lymphoma/Waldenström macroglobulinemia patients: a population-based study in Sweden. Blood. 2008; 112: 3052-6.
13. Kapoor P, Paludo J, Ansell S. Waldenström Macroglobulinemia: Familial predisposition and the role of genomics in prognosis and treatment selection. Curr. Treat. Options Oncol. 2016; 17: 16.
14. Treon SP, Hunter ZR, Aggarwal A y col. Characterization of familial Waldenström’s macroglobulinemia. Ann Oncol. 2006; 17: 488-94.
15. Steingrímsson V, Lund SH, Turesson I y col. Population-based study on the impact of the familial form of Waldenström macroglobulinemia on overall survival. Blood. 2015; 125: 2174-5.
16. Dimopoulos MA, Anagnostopoulos A. Waldenström's macroglobulinemia. Best Pract Res Clin Haematol. 2005; 18, 747-65.
17. Treon SP. How I treat Waldenström macroglobulinemia. Blood. 2009; 114:2375-85.
18. Kapoor P, Paludo J, Vallumsetla N, Greipp PR. Waldenström macroglobulinemia: what a hematologist needs to know. Blood Rev. 2015; 29:301-19.
19. Castillo JJ, Treon S. Initial evaluation of the patient with Waldenström Macroglobulinemia. Hematol Oncol Clin North Am. 2018; 32: 811-20.
20. San Miguel JF, Vidriales MB, Ocio E y col. Immunophenotypic analysis of Waldenström's macroglobulinemia. Semin Oncol. 2003; 30: 187-95.
21. Martín-Jiménez P, García-Sanz R, Balanzategui A y col. Molecular characterization of heavy chain immunoglobulin gene rearrangements in Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Haematologica. 2007; 92: 635-42.
22. Varettoni M Zibellini S, Capello D y col. Clues to pathogenesis of Waldenström macroglobulinemia and immunoglobulin M monoclonal gammopathy of undetermined significance provided by analysis of immunoglobulin heavy chain gene rearrangement and clustering of B-cell receptors. Leuk Lymphoma. 2013; 54: 2485-9.
23. Gachard N, Parrens M, Soubeyran I y col. IGHV gene features and MYD88 L265P mutation separate the three marginal zone lymphoma entities and Waldenström macroglobulinemia/lymphoplasmacytic lymphomas. Leukemia. 2013; 27: 183-9.
24. Agathangelidis A, Darzentas N, Hadzidimitriou A y col. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012; 119: 4467- 75.
25. Braggio E, Keats JJ, Leleu X y col. Identification of copy number abnormalities and inactivating mutations in two negative regulators of nuclear factor-kappaB signaling pathways in Waldenström’s macroglobulinemia. Cancer Res. 2009; 69: 3579-88.
26. Braggio E, Fonseca R. Genomic abnormalities of Waldenström Macroglobulinemia and related low-grade B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2013; 13: 198-201.
27. Schop RF, Jalal SM, Van Wier SA y col. Deletions of 17p13.1 and 13q14 are uncommon in Waldenström macroglobulinemia clonal cells and mostly seen at the time of disease progression. Cancer Genet Cytogenet. 2002; 132: 55-60.
28. Rivera A, Li M, Beltran G, Krause J. Trisomy 4 as the sole cytogenetic abnormality in a Waldenström macroglobulinemia. Cancer Genet Cytogenet. 2002; 133: 172–3.
29. Terre C, Nguyen-Khac F, Barin C y col. Trisomy 4, a new chromosomal abnormality in Waldenström's macroglobulinemia: a study of 39 cases. Leukemia. 2006; 20: 1634-6.
30. Braggio E, Dogan A, Keats JJ y col. Genomic analysis of marginal zone and lymphoplasmacytic lymphomas identified common and disease-specific abnormalities. Mod Pathol. 2012; 25: 651-60.
31. Nguyen-Khac F, Lambert J, Chapiro E y col. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenström’s macroglobulinemia. Haematologica. 2013; 98: 649-54.
32. Schop R, Kuehl W, Van Wier S y col. Waldenström macroglobulinemia neoplastic cells lack immunoglobulin heavy chain locus translocations but have frequent 6q deletions. Blood. 2002; 100: 2996–3001.
33. Ocio EM, Schop RF, Gonzalez B, y col. 6q deletion in Waldenström macroglobulinemia is associated with features of adverse prognosis. Br J Haematol. 2007; 136: 80- 6.
34. Chang H, Qi C, Trieu Y y col. Prognostic relevance of 6q deletion in Waldenström's macroglobulinemia: a multicenter study. Clin Lymphoma Myeloma Leuk. 2009; 9: 36-8.
35. Hunter ZR, Xu L, Yang G y col. The genomic landscape of Waldenstöm’s Macroglobulinemia is characterized by highly recurring MYD88 and WHIM- like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014; 123: 1637-46.
36. Varettoni M, Zibellini S, Defrancesco I y col. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017; 102: 2077- 85.
37. Guerrera ML, Tsakmaklis N, Xu Ly col. MYD88 mutated and wild-type Waldenström’s Macroglobulinemia: Characterization of chromosome 6q gene losses and their mutual exclusivity with mutations in CXCR4. Haematologica. 2018; 103: e408-e11.
38. Sekiguchi N, Nomoto J, Nagata A y col. Gene expression profile signature of aggressive Waldenström Macroglobulinemia with chromosome 6q deletion. Biomed Res Int. 2018; 2018: 6728128.
39. Shaffer AL, Lin KI, Kuo TC y col. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002; 17: 51-62.
40. Hodge LS, Ziesmer SC, Yang ZZ y col. IL-21 in the bone marrow microenvironment contributes to IgM secretion and proliferation of malignant cells in Waldenström macroglobulinemia. Blood. 2012; 120: 3774-82.
41. Treon SP, Xu L, Yang G, Zhou Y y col. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med. 2012; 367: 826-33.
42. Xu L, Hunter ZR, Tsakmaklis N y col. Clonal architecture of CXCR4 WHIM- like mutations in Waldenström macroglobulinaemia. Br J Haematol. 2016; 172:735-44.
43. Castillo JJ, Hunter ZR, Yang G, Treon SP. Novel Approaches to targeting MYD88 in Waldenström Macroglobulinemia. Expert Rev Hematol. 2017; 10: 739-44.
44. Treon S, Cao Y, Xu L y col. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenström macroglobulinemia. Blood. 2014; 123: 2791-6.
45. Poulain S, Roumier C, Decambron A y col: MYD88 L265P mutation in Waldenström macroglobulinemia. Blood. 2013; 121: 4504-11.
46. Treon SP, Gustine J, Xu L y col: MYD88 wild-type Waldenström macroglobulinaemia: Differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018; 180: 374-80.
47. Xu L, Hunter Z, Guang Y y col. MYD88 L265P in Waldenström macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013; 121: 2051-8.
48. Capaldi I, May A, Schmitt-Graeff A y col. Detection of MYD88 L265P mutations in formalin-fixed and decalcified BM biopsies from patients with lymphoplasmacytic lymphoma. Exp Mol Pathol. 2014; 97: 57-65.
49. Xu L, Hunter ZR, Yang G y col. Detection of MYD88 L265P in peripheral blood of patients with Waldenström’s macroglobulinemia and IgM monoclonal gammopathy of undetermined significance. Leukemia. 2014; 28: 1698-704.
50. Gustine J, Meid K, Xu L, Hunter ZR, Castillo JJ, Treon SP. To select or not to select? The role of B- cell selection in determining the MYD88 mutation status in Waldenström macroglobulinaemia. Br J Haematol. 2017; 176: 822-4.
51. Drandi D, Genuardi E, Dogliotti I y col. Highly sensitive MYD88L265P mutation detection by droplet digital polymerase chain reaction in Waldenström macroglobulinemia. Haematologica. 2018; 103: 1029-37.
52. Greco A, Tedeschi A, Varettoni M y col.Factors predicting transformation of asymptomatic IgM monoclonal gammopathy. Clin. Lymphoma Myeloma Leuk. 2011; 11, 77-9.
53. Varettoni M, Arcaini L, Zibellini S y col. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenström's macroglobulinemia and related lymphoid neoplasms. Blood. 2013; 121, 2522-8.
54. Kyle RA, Benson JT, Larson DR y col. Progression in smoldering Waldenström macroglobulinemia: long-term results. Blood. 2012; 119: 4462- 6.
55. Yang G, Zhou Y, Liu X y col. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood. 2013; 122: 1222-32.
56. Roccaro AM, Sacco A, Jimenez C y col. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014; 123: 4120-31.
57. Hunter ZR, Yang G, Xu L y col. Genomics, signaling, and treatment of Waldenström macroglobulinemia. J Clin Oncol. 2017; 35: 994-1001.
58. Dotta L, Tassone L, Badolato R. Clinical and genetic features of warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome. Current Mol Med. 2011; 11, 317-25.
59. Ngo HT, Leleu X, Lee J y col. SDF-1/CXCR4 and VLA-4 interaction regulates homing in Waldenström macroglobulinemia. Blood. 2008; 112: 150-8.
60. Busillo J and Benovica J. Regulation of CXCR4 Signaling. Biochim Biophys Acta. 2007; 1768: 952-63.
61. Lagane B, Chow KYC, Balabanian K y col. CXCR4 dimerization and beta- arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008; 112: 34-44.
62. Schmidt J, Federmann B, Schindler N y col. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015; 69: 795-803.
63. Castillo JJ, Treon SP. Toward personalized treatment in Waldenström macroglobulinemia. Hematology Am Soc Hematol Educ Program. 2017; 2017: 365-70.
64. Jimenez C, Prieto-Conde MI, Garcia-Alvarez M y col. Unraveling the heterogeneity of IgM monoclonal gammopathies: a gene mutational and gene expression study. Ann Hematol. 2018; 97: 475–84.
65. Treon SP, Gustine J, Meid K y col: Ibrutinib monotherapy in symptomatic, treatment-naïve patients with Waldenström macroglobulinemia. J Clin Oncol. 2018; 36: 2755-61.
66. Xu L, Zachary R, Hunter N y col. Clonal architecture of CXCR4 WHIM-like mutations in Waldenström Macroglobulinemia. British J of Hematol. 2016; 172: 735-44.
67. Wu YY, Jla M, Cai H y col. Detection of MYD88L265P and CXCR4S338X mutations by cell-free DNA in Waldenström Macroglobulinemia. Annals of Hematology. 2020; 99: 1763-9.
68. Hunter ZR, Xu L, Yang G y col. Transcriptome sequencing reveals a profile that corresponds to genomic variants in Waldenström macroglobulinemia. Blood. 2016; 128: 827-38.
69. Castillo JJ, Moreno D, Arbelaez M, Hunter Z, Treon S. CXCR4 mutations affect presentation and outcomes in patients with Waldenström macroglobulinemia: A systematic review. Expert Rev Hematol. 2019; 12: 1-9.
70. Hunter Z, Yang G, Xu L, Liu X, Castillo J, Treon S. Genomics, Signaling, and Treatment of Waldenstrom Macroglobulinemia. J Clin Oncol. 2017; 35: 994-1001.
71. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015; 94:193-205.
72. Argyropoulos KV, Vogel R, Ziegler C y col. Clonal B cells in Waldenström’s macroglobulinemia exhibit functional features of chronic active B-cell receptor signaling. Leukemia. 2016; 30: 1116–25.
73. Cao Y, Hunter ZR, Liu X y col. The WHIM-like CXCR4 (S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenström's Macroglobulinemia. Leukemia. 2015; 29: 169-76.
74. Treon SP, Tripsas, CK, Meid K y col. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N Eng J Med. 2015; 372: 1430-40.
75. Treon SP, Xu L, Hunter Z. MYD88 Mutations and response to ibrutinib in Waldenström’s macroglobulinemia. N Eng J Med. 2015; 373: 584-6.
76. Dimopoulos MA, Trotman J, Tedeschi A y col. Ibrutinib for patients with rituximab-refractory Waldenström’s macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol. 2017; 18:241–50.
77. Castillo JJ, Xu L, Gustine J y col. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. Br J Haematol. 2019; 187:356-3.